




Sarkome – Knochentumoren – Weichteilkrebs Allgemeine Informationen
Knochenkrebs / Knochentumor: Primäre Knochentumoren bzw. Knochenkrebs sind seltene Krebsarten, die in den Knochen entstehen. Im Vereinigten Königreich (mit 67 Millionen Einwohnern) werden jährlich etwa 550 Menschen mit einem Knochentumor oder Knochenkrebs diagnostiziert (etwa 8 pro 1 Million).
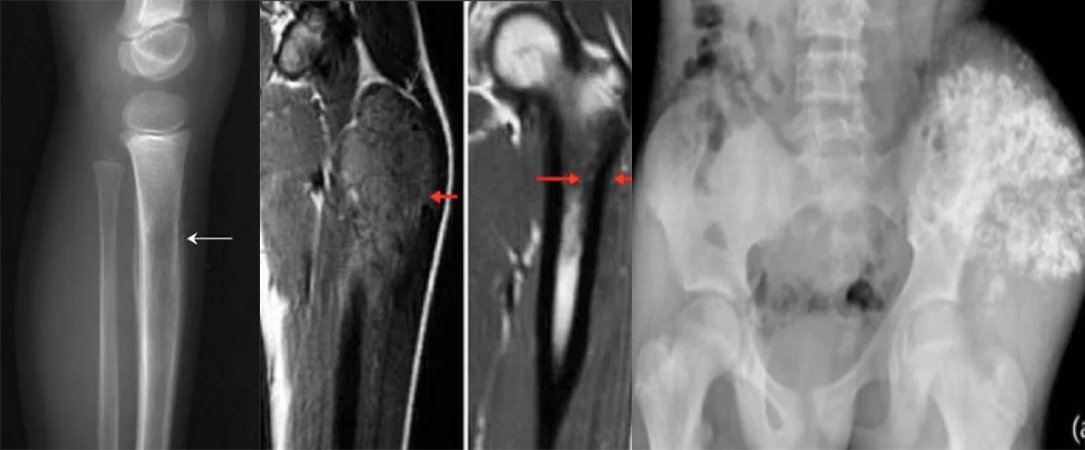
Symptome von Knochentumoren / Knochenkrebs: Obwohl Knochenkrebs grundsätzlich jeden Knochen befallen kann, tritt er meist in den langen Röhrenknochen der Arme und Beine auf. Zu den Hauptsymptomen gehören:
- Anhaltende, zunehmend starke Knochenschmerzen, die auch nachts bestehen
- Schwellung und Rötung (bei Gelenknähe Bewegungseinschränkung möglich)
- Tastbarer Knoten am Knochen
- Geschwächter Knochen, der leichter als gewöhnlich bricht
- Hinken
Wenn Sie oder Ihr Kind anhaltende, starke oder zunehmende Knochenschmerzen haben, sollten Sie einen orthopädischen Onkologen aufsuchen.
Was ist ein Knochentumor? Primärer Knochentumor bzw. Knochenkrebs ist eine seltene Krebsart, die in den Knochen beginnt. Im Vereinigten Königreich werden jährlich etwa 550 neue Fälle diagnostiziert (8 pro 1 Million Einwohner).
Genesung nach der Operation bei Knochentumoren – Was erwartet Sie?
Jede Operation ist individuell. Sie sollten Ihr Behandlungsteam fragen, was Sie direkt nach dem Eingriff erwartet. In der Regel gilt:
Sie wachen im Aufwachraum auf und werden dort von medizinischem Personal überwacht. Sobald Sie wach und stabil sind, werden Sie auf ein normales Krankenzimmer verlegt.
Sie haben einen großen Verband, der Ihre Beweglichkeit einschränken kann. Dieser bleibt einige Tage bis Wochen bestehen.
Plastische Drainagen können gelegt sein. Die Pflegekräfte leeren diese regelmäßig. Sie werden entfernt, sobald die Wundflüssigkeit versiegt.
Sie haben Schmerzen und erhalten Schmerzmittel – meist intravenös. Es kann auch eine PCA (Patientengesteuerte Analgesie) verwendet werden, bei der Sie selbst per Knopfdruck dosieren.
Sie erhalten Flüssigkeit über eine Infusion, bis Sie wieder selbstständig essen und trinken können. Meist ist dies innerhalb von 6 Stunden nach der OP möglich.
Zur Infektionsvorbeugung werden Antibiotika verabreicht – am OP-Tag und manchmal 1–2 Tage danach.
Gegebenenfalls wird ein Blasenkatheter eingesetzt, um den Urin abzuleiten.
Nach der OP kann eine zusätzliche Behandlung wie Chemotherapie oder Strahlentherapie notwendig sein, um verbleibende Krebszellen zu bekämpfen. Diese sogenannte adjuvante Therapie beginnt meist einige Wochen nach der Wundheilung.
Die Erholungszeit variiert individuell und hängt von der Art der Operation ab. Der Krankenhausaufenthalt dauert meist 2–7 Tage.
Die Hautwunde heilt in der Regel innerhalb von 2 Wochen. Bis der Knochen vollständig verheilt ist, kann es 1–2 Monate dauern. In einigen Fällen werden nicht lebende Implantate oder Prothesen eingesetzt, um die nach der Tumorentfernung entstandene Lücke zu füllen – jedoch kann es langfristig zu Infektionen, Lockerungen oder Wundheilungsstörungen kommen.
Zur Vermeidung solcher Komplikationen bevorzugen wir die Verwendung von lebendem Knochengewebe (Knochentransplantationen), auch wenn diese Eingriffe schwieriger sind und die Heilung länger dauert. Denn Metall kann lebendes Knochengewebe niemals vollständig ersetzen.
Nach lebenden Knochentransplantationen kann es bis zu 1 Jahr dauern, bis Knochenheilung und vollständige Belastbarkeit erreicht sind. Dennoch ist dies ein Verfahren, das wir langfristig bevorzugen, da es lebendes Gewebe nutzt.
Bei manchen Patient:innen können jedoch auch interne Prothesen anstelle von Knochentransplantaten verwendet werden. Eine sorgfältige Rehabilitation nach gliedmaßenerhaltender Operation ist entscheidend, um die Funktionalität von Arm oder Bein wiederherzustellen.
Falls Komplikationen auftreten, können weitere Eingriffe erforderlich werden. Bei Kindern kann eine zusätzliche Operation notwendig sein, um das Wachstum der gegenüberliegenden Gliedmaße auszugleichen.
Behandlung von Knochentumoren: In der Behandlung kombinieren wir Operation, Strahlentherapie, Chemotherapie sowie gezielte Therapien (z. B. Tyrosinkinase-Inhibitoren). Der Behandlungsplan wird individuell angepasst – je nach Tumorart, Lokalisation, Stadium und weiteren Faktoren.
Gezielte Therapie („Smart Drugs“): Durch die bessere Kenntnis genetischer Veränderungen bei Knochentumoren wurden neue Medikamente entwickelt, die gezielt auf diese Mechanismen wirken. Diese unterscheiden sich in ihrer Wirkung und ihren Nebenwirkungen von der klassischen Chemotherapie. Sie kommen vor allem bei Tumoren wie Chordomen zum Einsatz, bei denen Chemotherapie wenig wirksam ist. Andere medikamentöse Optionen sind knochengerichtete Therapien und Immuntherapien. Zielgerichtete Medikamente (z. B. Kinasehemmer) blockieren Signale in der Zelle, die das Tumorwachstum fördern. Dadurch wird das Wachstum bestimmter Tumore gestoppt oder verlangsamt – z. B. bei Chordomen oder fortgeschrittenem Chondrosarkom.
- Imatinib
- Dasatinib
- Sunitinib
- Erlotinib
- Lapatinib
- Sorafenib
- Regorafenib
- Pazopanib
Diese Medikamente werden in Tablettenform ein- bis zweimal täglich eingenommen. Zu den Nebenwirkungen zählen Übelkeit, Muskelschmerzen und Müdigkeit.
Denosumab ist ein RANKL-Inhibitor. Obwohl es in den letzten Jahren von einigen Fachgesellschaften zur Behandlung von Riesenzelltumoren empfohlen wurde, haben neuere Studien gezeigt, dass es zur bösartigen Entartung von Riesenzelltumoren beitragen kann. Daher hat es an Beliebtheit verloren. Interferon alfa-2b kann bei wiederkehrenden Riesenzelltumoren oder bei Metastasen eingesetzt werden. Zu den Nebenwirkungen dieser Therapie zählen grippeähnliche Muskelschmerzen, Knochenschmerzen, Fieber, Kopfschmerzen, Müdigkeit, Übelkeit und Erbrechen.
Was ist ein Sarkom? Symptome eines Sarkoms – Was ist Knochenkrebs?
Ein Sarkom ist ein bösartiger Tumor, also eine Krebsart, die aus mesenchymalen Zellen (Bindegewebe) hervorgeht. Der Begriff Bindegewebe umfasst Knochen, Knorpel, Fett, Gefäße und weitere strukturelle Gewebe. Sarkome können sich in all diesen Gewebearten entwickeln. Entsprechend gibt es viele Untertypen von Sarkomen, die nach der Herkunft des betroffenen Gewebes oder Zelltyps klassifiziert werden. Sarkome sind primäre Tumoren des Bindegewebes – sie entstehen also direkt dort. Dies unterscheidet sie von sekundären (metastasierten) Tumoren, bei denen sich der Krebs aus einem anderen Organ (z. B. Lunge, Brust oder Prostata) auf das Bindegewebe ausgebreitet hat. Sarkome gehören zu den fünf Hauptkrebsarten, die nach dem Zelltyp, aus dem sie entstehen, eingeteilt werden. Das Wort „Sarkom“ stammt vom griechischen σάρκωμα (sarōma) – „fleischige Wucherung“, von σάρξ (sarx) – „Fleisch“.
Sarkomklassifikation – Sarkomarten:
Sarkome werden in zwei Hauptgruppen unterteilt: Knochensarkome und Weichteilsarkome. Jede dieser Gruppen umfasst wiederum verschiedene Subtypen. In den USA veröffentlicht das American Joint Committee on Cancer (AJCC) Leitlinien zur Klassifikation dieser Untergruppen.
Subtypen der Knochensarkome:
- Osteosarkom
- Chondrosarkom
- Schlecht differenzierte Rund-/Spindelzelltumoren (einschließlich Ewing-Sarkom)
- Hämangioendotheliom
- Angiosarkom
- Fibrosarkom / Myofibrosarkom
- Chordom
- Adamantinom
- Weitere:
- Liposarkom
- Leiomyosarkom
- Bösartiger Tumor der peripheren Nervenscheide
- Rhabdomyosarkom
- Synovialsarkom
- Bösartiger solitärer fibröser Tumor
Subtypen der Weichteilsarkome: Was ist Weichteilkrebs?
- Liposarkom (Liposarkom-Krebs) (einschließlich: atypischer lipomatöser Tumor / gut differenziertes Liposarkom, undifferenziertes Liposarkom, myxoides Liposarkom, pleomorphes Liposarkom, und myxoides pleomorphes Liposarkom)
- Atypischer lipomatöser Tumor
- Dermatofibrosarkoma protuberans (einschließlich pigmentierter Varianten)
- Dermatofibrosarkoma protuberans, fibrosarkomatös
- Riesenzell-Fibroblastom
- Bösartiger solitärer fibröser Tumor
- Entzündlicher myofibroblastischer Tumor
- Myofibroblastisches Sarkom niedrigen Grades
- Fibrosarkom (einschließlich adultem und sklerosierend-epithelioidem Typ)
- Myxofibrosarkom (ehemals myxoides malignes fibröses Histiozytom)
- Fibromyxoides Sarkom niedrigen Grades
- Riesenzelltumor des Weichgewebes
- Leiomyosarkom
- Bösartiger Glomustumor
- Rhabdomyosarkom (einschließlich: embryonal, alveolär, pleomorph und spindelzellig/sklerosierend)
- Hämangioendotheliom (einschließlich: retiform, pseudomyogen und epitheloidzellig)
- Weichteilangiosarkom
- Extraskelettales Osteosarkom
- Maligner gastrointestinaler Stromatumor (GIST)
- Bösartiger Tumor der peripheren Nervenscheide (einschließlich epitheloider Variante)
- Maligner Triton-Tumor
- Bösartiger granulärer Zelltumor
- Maligner ossifizierender fibromyxoider Tumor
- Stromales Sarkom (wenn nicht anders angegeben)
- Myoepitheliales Karzinom
- Maligner phosphaturischer mesenchymaler Tumor
- Synovialsarkom (einschließlich: spindelzellig, biphasisch und nicht näher bezeichnet)
- Epitheloidzelliges Sarkom
- Alveoläres Weichteilsarkom
- Klarzelliges Sarkom des Weichgewebes
- Extraskelettales myxoides Chondrosarkom
- Extraskelettales Ewing-Sarkom
- Interdigitiertes dendritisches Zellsarkom
- Desmoplastischer Tumor kleiner rundzellige Zellen
- Extrarenales Rhabdoid-Tumor
- Perivaskulärer epitheloidzelliger Tumor (nicht näher bezeichnet)
- Intimales Sarkom
- Undifferenziertes spindelzelliges Sarkom
- Undifferenziertes pleomorphes Sarkom
- Undifferenziertes rundzelliges Sarkom
- Undifferenziertes epitheloides Sarkom
- Undifferenziertes Sarkom (wenn nicht anders angegeben)
Anzeichen und Symptome eines Sarkoms: Symptome eines Knochentumors: Symptome eines Knochenkrebses: Symptome eines Liposarkoms:
Zu den Symptomen von Knochensarkomen zählen typischerweise Knochenschmerzen, insbesondere nachts, und Schwellungen um die Tumorstelle herum.
Symptome von Weichteilkrebs im Bein: Symptome von Weichteilkrebs:
Die Symptome von Weichteilsarkomen variieren, sie treten jedoch meist als harte, oft schmerzlose Knoten oder Knötchen auf. Gastrointestinale Stromatumoren (GIST, ein Subtyp des Weichteilsarkoms) sind oft asymptomatisch, können aber mit unklaren Beschwerden wie Bauchschmerzen, Darmblutungen, Völlegefühl oder anderen Anzeichen eines Darmverschlusses einhergehen.
Sarkomursachen und Risikofaktoren:
Die Ursache der meisten Knochensarkome ist unbekannt, jedoch werden mehrere Faktoren mit einem erhöhten Risiko für die Entwicklung eines Knochensarkoms in Verbindung gebracht. Einer dieser Risikofaktoren ist eine frühere Exposition gegenüber ionisierender Strahlung (z. B. eine vorherige Strahlentherapie). Therapeutische Strahlung wird nach 10 bis 20 Jahren mit Sarkomen in Verbindung gebracht. Die Exposition gegenüber alkylierenden Substanzen, die in manchen Chemotherapeutika gegen Krebs enthalten sind, erhöht ebenfalls das Risiko für Knochensarkome. Bestimmte vererbte genetische Syndrome wie das Li-Fraumeni-Syndrom, erbliche Mutationen des RB1-Gens und die Paget-Knochenkrankheit werden mit einem erhöhten Risiko für die Entwicklung eines Knochensarkoms in Verbindung gebracht. Die meisten Weichteilsarkome werden durch das verursacht, was Ärzte als „sporadische“ (oder zufällige) genetische Mutationen in den Zellen der betroffenen Person bezeichnen. Es gibt jedoch einige Risikofaktoren, die mit einem erhöhten Risiko für die Entwicklung eines Weichteilsarkoms verbunden sind. Eine frühere Exposition gegenüber ionisierender Strahlung ist ein solcher Risikofaktor. Die Exposition gegenüber Vinylchlorid (z. B. Dämpfen bei der Herstellung von Polyvinylchlorid (PVC), Arsen und Thorotrast ist mit einem erhöhten Risiko für Angiosarkom verbunden. [2][3] Lymphödeme infolge bestimmter Brustkrebsbehandlungen sind ebenfalls ein Risikofaktor für die Entwicklung eines Angiosarkoms. Wie bei Knochensarkomen sind auch bestimmte vererbte genetische Syndrome, darunter das Li-Fraumeni-Syndrom, die familiäre adenomatöse Polyposis, die Neurofibromatose Typ 1 und erbliche Mutationen des RB1-Gens, mit einem erhöhten Risiko für die Entwicklung eines Weichteilsarkoms verbunden. Das Kaposi-Sarkom wird durch das Kaposi-Sarkom-assoziierte Herpesvirus (HHV-8) verursacht.
Sarkommechanismen:
Die genauen molekularen Veränderungen, die zu einem Sarkom führen, sind nicht immer bekannt, aber bestimmte Sarkomarten werden mit bestimmten genetischen Mutationen in Verbindung gebracht. Beispiele: Die meisten Fälle von Ewing-Sarkomen sind mit einer Chromosomentranslokation verbunden, bei der ein Teil von Chromosom 11 mit einem Teil von Chromosom 22 fusioniert. Dies führt dazu, dass das EWSR1-Gen mit anderen Genen fusioniert, einschließlich des FLI1-Gens in 90 % der Ewing-Fälle und des ERG-Gens in 5-10 % der Fälle. Diese Fusionen führen zur Produktion abnormer Proteine, aber es ist nicht genau bekannt, wie diese abnormen Proteine zu Krebs führen. Dermatofibrosarcoma protuberans ist oft mit einer Chromosomentranslokation verbunden, bei der das COL1A1-Gen mit dem PDGFRB-Gen fusioniert wird. Dies führt zu einer überaktiven PDGF-Signalgebung, die vermutlich die Zellteilung fördert und letztendlich zur Tumorentwicklung führt. Entzündliche myofibroblastische Tumoren gehen häufig mit einer Umlagerung des ALK-Gens und manchmal mit einer Umlagerung des HMGA2-Gens einher. Tenosynoviale Riesenzelltumoren (kein Sarkom, sondern ein nicht metastasierender und lokal aggressiver Weichteiltumor) gehen häufig mit einer chromosomalen Translokation zwischen Chromosom 1 und Chromosom 2 einher, wo sich das CSF1-Gen mit dem COL6A3-Gen verbindet. Dies führt zu einer erhöhten Produktion des CSF1-Proteins, das vermutlich eine Rolle bei der Krebsentstehung spielt. Viele Liposarkome gehen mit einer Amplifikation eines Teils des Chromosoms 12 einher, was zu zusätzlichen Kopien bekannter krebsfördernder Gene („Onkogene“) wie dem CDK4-Gen, dem MDM2-Gen und dem HMGA2-Gen führt.
Sarkomdiagnose: Knochensarkome:
Die Diagnose von Knochensarkomen beginnt mit einer ausführlichen Anamnese und körperlichen Untersuchung, die charakteristische Anzeichen und Symptome zutage fördern kann (siehe Anzeichen und Symptome oben). Einige Knochensarkome (wie das Osteosarkom) können mit erhöhten alkalischen Phosphatasewerten verbunden sein, andere (wie das Ewing-Sarkom) können mit einer erhöhten Blutsenkungsgeschwindigkeit einhergehen, doch Laboruntersuchungen sind für die Diagnose nicht besonders hilfreich. Wichtig ist jedoch, dass keines dieser Laborbefunde spezifisch für Knochensarkome ist. Dies bedeutet, dass erhöhte Laborwerte neben Sarkomen auch mit vielen anderen Erkrankungen in Verbindung gebracht werden und daher für eine definitive Diagnose eines Sarkoms nicht herangezogen werden können. Bildgebende Verfahren sind für die Diagnose entscheidend, und die meisten Ärzte werden zunächst eine Röntgenaufnahme anordnen. Andere bildgebende Verfahren, die häufig zur Diagnose eingesetzt werden, sind Magnetresonanztomographie (MRT) und Radioisotopen-Knochenszintigraphien. Die CT wird üblicherweise nicht zur Diagnose der meisten Knochensarkome eingesetzt, ist aber ein wichtiges Instrument zur Stadienbestimmung (siehe unten). Die endgültige Diagnose erfordert eine Biopsie des Tumors und die sorgfältige Untersuchung der Biopsieprobe durch einen erfahrenen Pathologen.
Weichteilsarkome: Weichteilsarkom: Weichteilkrebs: Was ist ein Weichteiltumor?
Die Diagnose von Weichteilsarkomen beginnt mit einer ausführlichen Anamnese und körperlichen Untersuchung. Bildgebende Verfahren können CT oder MRT umfassen, wobei die CT bei Weichteilsarkomen im Thorax, Abdomen oder Retroperitoneum bevorzugt wird. Auch die Positronen-Emissions-Tomographie (PET) kann zur Diagnose hilfreich sein, wird aber meist zur Stadienbestimmung eingesetzt (siehe unten). Wie bei Knochensarkomen erfordert die endgültige Diagnose eine Biopsie des Tumors mit histologischer Auswertung durch einen ausgebildeten Pathologen.
Sarkom-Staging:
Im Allgemeinen bezieht sich die Krebsstadienbestimmung darauf, wie weit der Krebs fortgeschritten ist, und basiert in der Regel auf Faktoren wie der Größe des Tumors und ob er sich auf andere Körperteile ausgebreitet hat. Die Stadienbestimmung ist wichtig, da das Stadium die Prognose (wahrscheinlicher Ausgang) sowie die Art der Behandlungen beeinflusst, die voraussichtlich gegen den Krebs wirksam sind. Bei der Stadienbestimmung von Sarkomen wird festgestellt, ob der Tumor in das umliegende Gewebe eingewachsen ist („lokale Invasion“), ob er sich auf die Lymphknoten ausgebreitet hat (Bildung von „Knotenmetastasen“) oder ob er sich auf andere Gewebe oder Organe im Körper ausgebreitet hat (Bildung von „Fernmetastasen“). Die gebräuchlichsten bildgebenden Verfahren zur Stadienbestimmung von Knochensarkomen sind MRT oder CT zur Beurteilung des Primärtumors, eine kontrastmittelverstärkte Thorax-CT zur Beurteilung, ob sich der Krebs in die Lunge ausgebreitet (d. h. metastasiert) hat, und eine Radioisotopen-Knochenszintigraphie zur Beurteilung, ob er sich auf andere Knochen ausgebreitet hat. Zur Stadienbestimmung bei Weichteilsarkomen gehört typischerweise eine MRT- oder CT-Aufnahme des Primärtumors zur Bestimmung der Tumorgröße sowie eine kontrastmittelverstärkte Thorax-CT zur Beurteilung metastasierter Tumoren in der Lunge.
Grad – Sarkomgrad:
Wie bei einigen anderen Krebsarten wird auch bei Sarkomen der Grad (niedrig, mittel oder hoch) anhand des Erscheinungsbilds der Tumorzellen unter dem Mikroskop bestimmt. Im Allgemeinen gibt der Grad an, wie aggressiv der Krebs ist und wie wahrscheinlich es ist, dass er sich in andere Körperteile ausbreitet („metastasiert“). Niedriggradige Sarkome haben eine bessere Prognose als hochgradige Sarkome und werden in der Regel operativ behandelt, obwohl manchmal auch Strahlentherapie oder Chemotherapie zum Einsatz kommen. Mittel- und hochgradige Sarkome werden häufiger mit einer Kombination aus Operation, Chemotherapie oder Strahlentherapie behandelt. Da höhergradige Tumoren eher metastasieren (invasiv werden und sich in lokale und entfernte Bereiche ausbreiten), werden sie aggressiver behandelt. Die Erkenntnis, dass viele Sarkome empfindlich auf Chemotherapie reagieren, hat die Überlebensrate der Patienten deutlich erhöht. Beispielsweise lag die Langzeitüberlebensrate von Kindern mit lokalisiertem Osteosarkom vor der Chemotherapie bei nur etwa 20 %, heute ist sie auf 60–70 % gestiegen.
Sarkombehandlung: Knochentumorchirurgie: Knochenkrebsbehandlung: Liposarkombehandlung:
Was ist ein Liposarkom: Sarkomkrebs: Weichteiltumor: Weichteilsarkome:
Bei den meisten Sarkomen, die sich nicht auf andere Körperteile ausgebreitet haben, ist eine Operation die häufigste Behandlungsmethode, und bei den meisten Sarkomen ist sie die einzige kurative Behandlung. Im Gegensatz zur Amputation wird heute bei mindestens 90 % der Extremitätensarkomfälle (Arm oder Bein) eine gliedmaßenerhaltende Operation durchgeführt, um die Gliedmaßen der Patienten zu erhalten. Zusätzliche Behandlungen, wie Chemotherapie (einschließlich Protonentherapie) oder Strahlentherapie (auch „Radiotherapie“ genannt), können vor der Operation (sogenannte „neoadjuvante“ Chemotherapie oder Radiotherapie) oder nach der Operation (sogenannte „adjuvante“ Chemotherapie oder Radiotherapie) verabreicht werden. Der Einsatz einer neoadjuvanten oder adjuvanten Chemotherapie und Strahlentherapie verbessert die Prognose vieler Sarkompatienten erheblich. Die Behandlung kann langwierig und schwierig sein und bei vielen Patienten etwa ein Jahr dauern. Die Behandlung eines Liposarkoms besteht üblicherweise aus einer chirurgischen Resektion, und je nach Aggressivität des Sarkoms wird eine Chemotherapie in Betracht gezogen. Strahlentherapie kann auch vor oder nach der chirurgischen Entfernung eines Liposarkoms eingesetzt werden. Pädiatrisches Rhabdomyosarkom wird üblicherweise mit Chemotherapie, Operation und manchmal Strahlentherapie behandelt. Die Langzeitüberlebensrate bei pädiatrischen Rhabdomyosarkompatienten liegt bei 50-85 %. Osteosarkom ist ein Knochenkrebs, der durch chirurgische Entfernung des Krebses, üblicherweise kombiniert mit Chemotherapie, behandelt wird. Strahlentherapie ist eine hilfreiche Alternative, wenn auch nicht so erfolgreich wie eine Operation. Früher glaubte man, dass höhere Chemotherapiedosen die Überlebenschancen erhöhen könnten. Hohe Chemotherapiedosen stoppen jedoch die Produktion von Blutzellen im Knochenmark und können schädlich sein. Stammzellen, die Menschen vor einer Hochdosis-Chemotherapie entnommen wurden, können dem Menschen zurücktransplantiert werden, wenn seine Blutzellzahl zu niedrig wird; dies wird als autologe hämatopoetische Stammzelltransplantation oder Hochdosisbehandlung mit Stammzellrettung bezeichnet. Bei der Untersuchung, ob eine autologe hämatopoetische Stammzelltransplantation nach einer Hochdosis-Chemotherapie günstiger war als eine Standarddosis-Chemotherapie, wurde kein signifikanter Unterschied festgestellt.
Verlauf der Lebenszeitprognose bei Sarkomen:
Das AJCC hat mehrere Faktoren identifiziert, die die Prognose von Knochensarkomen beeinflussen:
- Tumorgröße: Größere Tumoren haben tendenziell eine schlechtere Prognose als kleinere Tumoren.
- Tumorausbreitung in umliegendes Gewebe: Tumoren, die lokal in umliegendes Gewebe streuen, haben tendenziell eine schlechtere Prognose als Tumoren, die sich nicht über ihren Ursprungsort hinaus ausgebreitet haben.
- Stadium und Vorhandensein von Metastasen: Tumoren, die sich in Lymphknoten (selten bei Knochensarkomen) oder andere Organe oder Gewebe (z. B. die Lunge) ausgebreitet haben, haben eine schlechtere Prognose als Tumoren, die sich nicht ausgebreitet haben.
- Tumorgrad: Hochgradige Tumoren (Grad 2 und 3) haben tendenziell eine schlechtere Prognose als niedriggradige (Grad 1) Tumoren.
- Skelettale Lokalisation: Tumoren, die von der Wirbelsäule oder den Beckenknochen ausgehen, haben tendenziell eine schlechtere Prognose als Tumoren, die von den Arm- oder Beinknochen ausgehen.
Faktoren, die die Prognose bei Weichteilsarkomen außer GIST beeinflussen, sind:
- Stadium: Wie bei Knochensarkomen haben metastasierte Tumoren eine schlechtere Prognose als nicht metastasierte Tumoren.
- Grad: Die AJCC empfiehlt die Verwendung eines Grading-Systems für Weichteilsarkome, genannt „Grade“ der French Federation of Cancer Centers Sarcoma Group (FNCLCC). Hochgradige Tumoren haben eine schlechtere Prognose als niedriggradige Tumoren.
Der Hauptfaktor, der die Prognose bei GIST beeinflusst:
- Mitoserate: Die Mitoserate bezieht sich auf den Anteil der Zellen, die sich innerhalb des Tumors aktiv teilen. GISTs mit einer hohen Mitoserate haben eine schlechtere Prognose als GISTs mit einer niedrigen Mitoserate.
Sarkom-Überleben:
Laut Daten des US-amerikanischen National Cancer Institute (NCI) beträgt die 5-Jahres-Gesamtüberlebensrate bei Knochensarkomen 66,9 %. Die American Cancer Society (ACS) schätzt, dass im Jahr 2023 in den USA 2.140 Menschen an Knochensarkomen sterben werden, was 0,3 % aller krebsbedingten Todesfälle entspricht. Das durchschnittliche Sterbealter liegt bei 61 Jahren, der Tod kann jedoch in jeder Altersgruppe eintreten. Demnach ereignen sich 12,3 % der Todesfälle durch Knochensarkom bei Menschen unter 20 Jahren, 13,8 % bei Menschen zwischen 20 und 34 Jahren, 5,5 % bei Menschen zwischen 35 und 44 Jahren und 9,3 % bei Menschen über 45 Jahren. Bei Personen im Alter von 54 Jahren tritt das 5-Jahres-Überleben bei 13,5 % der 55- bis 64-Jährigen, 16,2 % der 65- bis 74-Jährigen, 16,4 % der 75- bis 84-Jährigen und 13,1 % der 85-Jährigen und Älteren auf. Die 5-Jahres-Gesamtüberlebensrate bei Weichteilsarkomen (unabhängig vom Stadium) beträgt 64,5 %, wobei die Überlebensrate von vielen Faktoren, einschließlich des Stadiums, beeinflusst wird. So beträgt die 5-Jahres-Überlebensrate bei Weichteilsarkomen, die sich nicht über den Primärtumor hinaus ausgebreitet haben („lokalisierte“ Tumoren), 80,8 % bei Weichteilsarkomen, die sich nur auf nahegelegene Lymphknoten ausgebreitet haben, 58,0 % und bei Weichteilsarkomen, die sich auf entfernte Organe ausgebreitet haben, 16,4 %. Die ACS schätzt, dass im Jahr 2023 5.140 Menschen an Weichteilsarkomen sterben werden, was 0,9 % aller Krebstodesfälle entspricht.
Epidemiologie:
Sarkome sind seltene Krebserkrankungen. Das Risiko, bei einem zuvor gesunden Menschen neu an Knochenkrebs zu erkranken, liegt unter 0,001 %, während das Risiko, neu an Weichteilsarkomen zu erkranken, zwischen 0,0014 und 0,005 % liegt. Die American Cancer Society schätzt, dass im Jahr 2023 in den USA 3.970 neue Fälle von Knochensarkomen und 13.400 neue Fälle von Weichteilsarkomen auftreten werden. Angesichts der geschätzten Gesamtzahl neuer Krebsdiagnosen (alle Krebsarten) von 1.958.310 bedeutet dies, dass Knochensarkome nur 0,2 % aller neuen Krebsdiagnosen ausmachen (und damit die 30. häufigste Krebsart) und Weichteilsarkomen nur 0,7 % aller neuen Krebsdiagnosen ausmachen (und damit die 22. häufigste Krebsart). Sarkome betreffen Menschen jeden Alters. Etwa 50 % der Knochensarkome und 20 % der Weichteilsarkome werden bei Menschen unter 35 Jahren diagnostiziert. Einige Sarkome, wie das Leiomyosarkom, das Chondrosarkom und der gastrointestinale Stromatumor (GIST), kommen bei Erwachsenen häufiger vor als bei Kindern. Die meisten hochgradigen Knochensarkome, darunter das Ewing-Sarkom und das Osteosarkom, kommen bei Kindern und jungen Erwachsenen deutlich häufiger vor. Im Jahr 2016 berichteten Wissenschaftler von der Entdeckung eines Osteosarkomtumors in einem 1,6 bis 1,8 Millionen Jahre alten Fossil im Skelett der ausgestorbenen Homininenart Australopithecus sediba. Damit ist dies der älteste bekannte Fall von Krebs beim Menschen. Knochenkrebs und andere Krebsarten befallen Homininen und Menschen also seit etwa 2 Millionen Jahren. Die Schritte der Diagnose und Behandlung decken lediglich einen Zeitraum von 100 Jahren ab.
Preise für Knochentumoroperationen: Die Kosten für eine Knochentumoroperation variieren je nach Art und Lage des Tumors, der Anzahl der Spezialisten, die die Operation durchführen, dem Ausmaß der Schädigung und der Frage, ob gliedmaßenerhaltende mikrochirurgische Eingriffe durchgeführt werden oder nicht.
Tumorbehandlung am Bein: Tumoren im Bein werden unter Erhalt der Gefäße und Nerven oder bei Bedarf durch Transplantation entfernt.
