





Knochentumoren
Eine angemessene und wirksame Untersuchung von Knochentumoren durch den orthopädischen Tumorchirurgen und sein Team (orthopädischer Tumorradiologe, orthopädischer Tumorpathologe, Gefäßchirurg, Mikrochirurg mit ausreichender Rekonstruktionserfahrung, Strahlenonkologe und medizinischer Onkologe) ist entscheidend für eine adäquate Behandlung des Patienten. Andernfalls wird der Patient falsch behandelt. Eine fehlerhafte oder verzögerte Diagnose kann zu schweren Behinderungen oder sogar zum Tod führen.

Die Diagnose beginnt mit der ausführlichen Anamnese der Beschwerden des Patienten, einer gründlichen klinischen Untersuchung und einer klassischen Röntgenaufnahme. Gutartige Tumoren erscheinen in der Regel als Läsionen, die keine Beschwerden verursachen, geografische Merkmale aufweisen, sklerotische Ränder haben und keine Knochenzerstörung oder periostale Reaktion hervorrufen.

Bösartige Tumoren hingegen verursachen regionale Schmerzen und zeigen im Röntgenbild ein lythisches, infiltrierendes Wachstum mit unscharfen Begrenzungen bei intaktem Knochen. Eine Zerstörung der Knochenhaut und/oder eine periostale Reaktion (Abbildung 2) kann beobachtet werden.
In diesem Stadium sollte ein klassischer Orthopäde seinen Patienten an einen Spezialisten für orthopädische Tumorchirurgie überweisen. Fortgeschrittene Bildgebung und Biopsie sind Verfahren, über die Ihr Tumorarzt entscheidet.
Klinisches Bild von Knochentumoren
Die Klinik ist sehr unterschiedlich. Läsionen können bei Patienten zufällig entdeckt werden oder sich durch Symptome wie Schmerzen und Schwellungen äußern. Zum Beispiel können fibröse Dysplasie oder Osteochondromatose mit einer Knochenverbiegung einhergehen. Frakturen, Weichteilverdickungen oder Schwellungen können zusammen mit dem Tumor im Knochen auftreten. Schmerzhafte Läsionen werden meist bei aggressiven (gutartig, aber aggressiv) und potenziell bösartigen Tumoren beobachtet. Da das klinische Spektrum gutartiger und bösartiger Tumoren sehr breit ist, sollte die Diagnose von einem auf Tumorchirurgie spezialisierten Arzt gestellt werden. Fehlerhafte oder unzureichende Diagnosen und Behandlungen können dazu führen, dass ein Patient, der eigentlich nur eine einfache Nachsorge oder Therapie benötigt, unnötigen, teuren und belastenden Untersuchungen und Behandlungen ausgesetzt wird. Ebenso kann eine unzureichende Diagnose und Behandlung von aggressiven und bösartigen Tumoren zum Verlust von Gliedmaßen oder sogar zum Tod führen.
Klinisches Bild bei gutartigen Tumoren
Es kann zu mäßigen Schmerzen kommen, die durch Schmerzmittel gelindert werden. Die Schmerzen entwickeln sich langsam und können mit körperlicher Aktivität oder einem Trauma zusammenhängen. Beispielsweise sind nächtliche Schmerzen, die gut auf Schmerzmittel ansprechen, typisch für ein Osteoidosteom. Frakturen, die durch eine Bewegung entstehen, die normalerweise keinen Knochenbruch verursachen würde, werden als pathologische Frakturen bezeichnet. Bei einigen gutartigen Läsionen können pathologische Frakturen durch einmalige oder wiederholte Traumata auftreten. WENN DIE LÄSION ZUFÄLLIG ENTDECKT WIRD, WÄHREND DER PATIENT KEINE SCHMERZEN ODER SYMPTOME HAT, IST SIE HÖCHSTWAHRSCHEINLICH GUTARTIG.

Ein erfahrener Tumorchirurg kann anhand des Alters des Patienten, der Lage der Läsion und des Röntgenbildes erkennen, ob die Läsion gutartig ist oder ob eine Biopsie erforderlich ist. Die radiologische Untersuchung sollte den gesamten Knochen umfassen. Gutartige Tumoren sind typischerweise geografisch begrenzt, haben schmale Übergangszonen und sklerotische Ränder (Abbildung 1). Eine endostale Ausdünnung kann auftreten, aber eine Zerstörung der Kortikalis ist selten. Ein weiteres hilfreiches Kriterium bei der Diagnose ist die Matrix der Läsion.
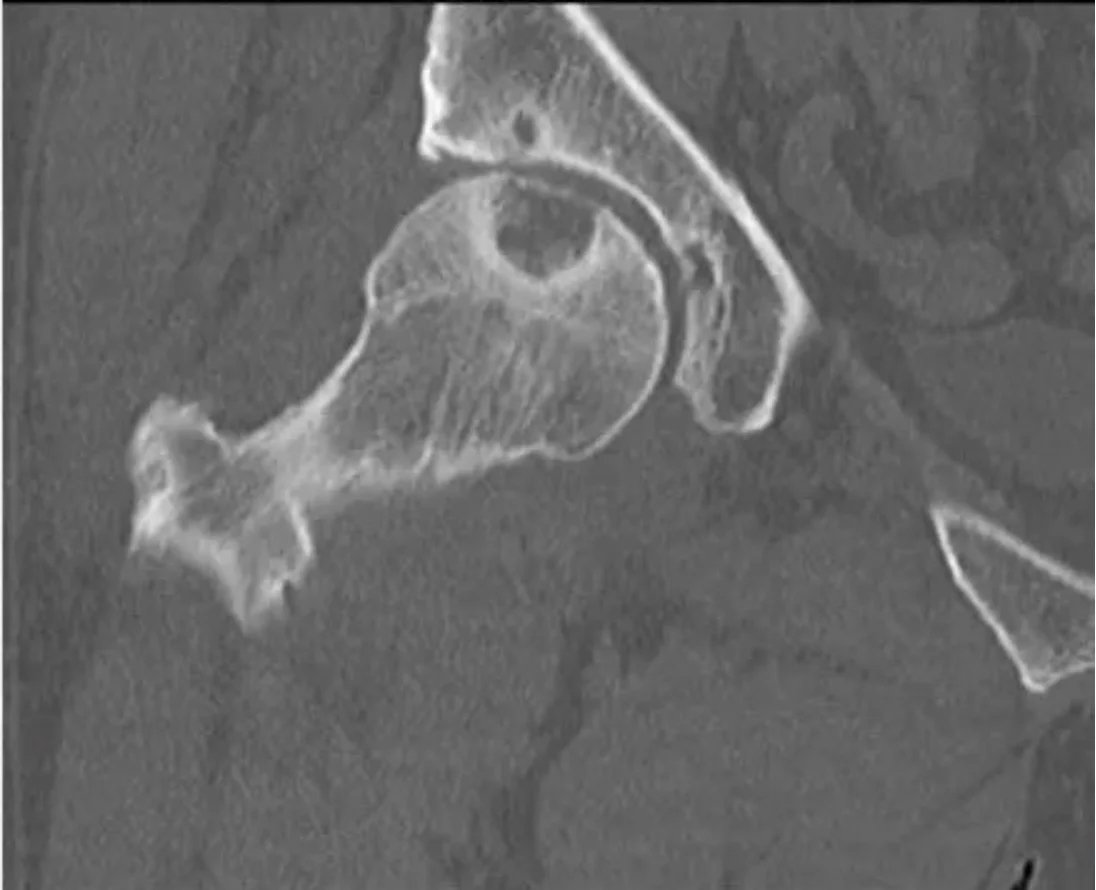
MRT zeigt die Weichteilkomponente, die Kontrastmittelanreicherung, das Knochenmarködem und die Signalcharakteristik der Läsion. Die Szintigrafie ist hilfreich bei der Beurteilung multipler Knochenbeteiligungen, wie bei polyostotischer fibröser Dysplasie, multipler Enchondromatose und Histiozytose. Eine „kalte“ Szintigrafie schließt eine aggressive oder maligne Läsion nicht aus (z. B. multiples Myelom, Metastasen eines Nierenzellkarzinoms). Degenerative Veränderungen und Überlastungsläsionen in gelenknahen Bereichen müssen ebenfalls in die Differenzialdiagnose einbezogen werden.
Wenn eine Zerstörung der Kortikalis, ein permeatives Wachstum und eine periostale Reaktion vorliegen, ist eine weiterführende Untersuchung unerlässlich. Wenn trotz CT und MRT keine eindeutige Diagnose gestellt werden kann, ist eine Biopsie erforderlich. Nach der Diagnose eines gutartigen Knochentumors kann in Intervallen von 3 bis 6 Monaten eine Beobachtung und Nachkontrolle durchgeführt werden, um die radiologische Stabilität zu überwachen. Ein chirurgischer Eingriff ist notwendig, um die Knochenzerstörung zu stoppen, pathologische Frakturen zu erkennen oder sich anbahnende Frakturen zu stabilisieren und Deformitäten zu verhindern.
Klinisches Bild bei bösartigen Tumoren
Das klinische Bild ist sehr unterschiedlich. Läsionen bei Patienten können zufällig entdeckt werden oder mit Symptomen wie Schmerzen und Schwellungen auftreten. Zum Beispiel zeigen Patienten mit fibröser Dysplasie oder Osteochondromatose häufig Anzeichen von Knochenverkrümmung. Die Schmerzen sind oft stark und Schmerzmittel reichen nicht aus. Es handelt sich um dumpfe und tiefliegende Schmerzen, die nicht mit Aktivität oder Ruhe in Zusammenhang stehen. Bei pathologischen Frakturen treten die Schmerzen plötzlich auf. Bei Tumoren an der Wirbelsäule oder im Kreuzbein können Taubheitsgefühle in Beinen und Füßen, Schwäche sowie Störungen der Blasen- und Darmfunktion auftreten. Eine begleitende Schwellung kann vorhanden sein. Müdigkeit, Schwäche und Fieber können ebenfalls vorkommen. Im Labor können erhöhte alkalische Phosphatasewerte, erhöhter Kalziumspiegel und Anämie festgestellt werden. Die familiäre Vorgeschichte von Krebs oder Sarkomen ist bei der Anamnese bedeutsam. Zum Beispiel haben das Retinoblastom, das Li-Fraumeni-Syndrom und das Rothmund-Thomson-Syndrom eine prädisponierende Wirkung auf Knochentumoren wie Osteosarkom. Es wurde berichtet, dass sich Läsionen bei multipler Enchondromatose verschlechtern können. Auch bei Patienten mit Paget-Krankheit, einer Vorgeschichte von Strahlentherapie, chronischer Osteomyelitis oder Knocheninfarkten kann sich ein Sarkom entwickeln. Die Biopsie sollte von einem orthopädischen Tumorchirurgen durchgeführt werden, da sie so geschlossen wie möglich erfolgen sollte, was Erfahrung erfordert. Zudem sollte die Biopsiestelle so gewählt werden, dass keine wichtigen Gefäße oder Nerven kontaminiert werden. Der Ort der Biopsie muss entlang der Linie liegen, die bei der eigentlichen Tumoroperation vom Tumorchirurgen bevorzugt wird. Andernfalls können aufwendige und belastende Operationen notwendig werden, wie z. B. erhebliche Muskel- und Gewebeverluste, gefährliche Gefäßtransplantationen, Nervenverluste und Hauttransplantationen. Dies kann das Risiko eines Gliedmaßenverlustes erhöhen und die Lebenserwartung verkürzen. Wird ein bösartiger Knochentumor diagnostiziert, muss ein Staging erfolgen. Dazu ist eine rasche PET-Bildgebung erforderlich. Bei Patienten über 40 Jahren handelt es sich bei der Läsion meist um metastasiertes Karzinom, multiples Myelom oder Lymphom. Zudem sollten bei Frauen die Brust und bei Männern die Prostata untersucht und eine Eiweißelektrophorese im Serum und Urin durchgeführt werden. Die Behandlung kann einen multidisziplinären Ansatz erfordern, unter Einbeziehung von Strahlen- und medizinischen Onkologen, unter der Leitung eines orthopädischen Tumorchirurgen. Frakturen, Weichteilgeschwülste und Schwellungen können zusammen mit dem Tumor im Knochen auftreten. Schmerzhaft sind in der Regel aggressive (gutartige, aber aggressive) und potenziell bösartige Tumoren. Da das klinische Spektrum gutartiger und bösartiger Tumoren sehr breit ist, sollte die Diagnose von einem in der Tumorchirurgie erfahrenen Arzt gestellt werden. Falsche und unzureichende Diagnosen und Behandlungen können dazu führen, dass ein Patient, der nur einer einfachen Nachsorge oder Behandlung bedarf, unnötigen, teuren und belastenden Untersuchungen und Therapien ausgesetzt wird. Ebenso kann eine unzureichende Diagnose und Behandlung von aggressiven und bösartigen Tumoren zum Verlust von Gliedmaßen oder sogar zum Tod führen.
Weichteiltumoren
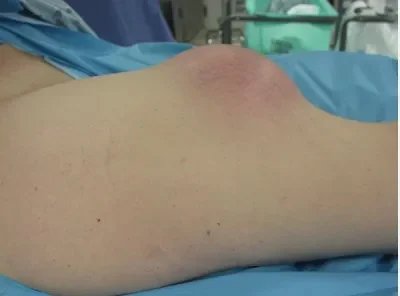
Weichteiltumoren kommen häufiger vor als Knochentumoren und sind meist gutartig. Der Diagnoseprozess beginnt mit einer ausführlichen Anamnese und Untersuchung. Röntgen, MRT, Ultraschall, CT und PET werden für weiterführende Untersuchungen eingesetzt. Diese Verfahren dienen auch der Beurteilung des Ansprechens auf die Behandlung. Gutartige Tumoren treten 100-mal häufiger auf als bösartige. Lage, Größe und Konsistenz der Raumforderung sind in der Anamnese und klinischen Untersuchung von Bedeutung. Auch ihre Tiefe im Verhältnis zur Faszie wird geprüft.
Fragen, die gestellt werden sollten:
- Wann wurde die Raumforderung erstmals bemerkt?
- Gab es eine Größenzunahme?
- Wie schnell wächst die Masse?
- Gab es zwischenzeitlich eine Größenabnahme?
- Gibt es weitere begleitende Raumforderungen?
- Treten Schmerzen, Hautrötung oder eine Schwellung der regionalen Lymphknoten auf?
- Liegt Fieber, Schüttelfrost oder Nachtschweiß vor?
Zusätzlich sollten Fragen zu Themen wie Trauma, Einnahme von Blutverdünnern, frühere Krebserkrankungen und Weltreisen gestellt werden.
EINE KLEINE, WEICHE, OBERFLÄCHLICHE UND SEIT JAHREN STABILE RAUMFORDERUNG IST SEHR WAHRSCHEINLICH GUTARTIG.
GROSSE, HARTE, TIEFLIEGENDE UND WACHSENDE MASSE HABEN EINE HOHE WAHRSCHEINLICHKEIT, BÖSARTIG ZU SEIN.
Bei Personen, die Blutverdünner einnehmen, können selbst bei leichten Traumata Hämatome entstehen. Diese Hämatome können jedoch eine zugrunde liegende Krebserkrankung überdecken.
Das Vorhandensein von Ossifikationen in der Raumforderung hilft bei der Differenzialdiagnose. Zum Beispiel tritt bei einer gutartigen, traumabedingten Raumforderung namens Myositis ossificans die Verkalkung peripher auf, während sie beim (bösartigen) Weichteilosteosarkom zentral liegt. Verkalkungen wirken unorganisierter als Ossifikationen. Phlebolithen können bei Gefäßmissbildungen auftreten, und dystrophe Verkalkungen finden sich in einigen Lipomen. Wenn eine solche dystrophe Verkalkung aggressiv erscheint, deutet dies auf ein Synovialsarkom hin – eine extrem bösartige Form. Wenn sich multiple, gleichmäßige Verkalkungen rund um das Gelenk befinden, sollte an einen gutartigen Tumor namens Synovialchondromatose gedacht werden. Eine aggressive periostale Reaktion oder Kortikaliszerstörung im angrenzenden Knochengewebe auf dem Röntgenbild weist mit hoher Wahrscheinlichkeit auf eine bösartige Raumforderung hin. Ein hohes T2-Signal im MRT, eine Masse größer als 3 cm, unterhalb der Faszie gelegen, peripheres Ödem, Blutung innerhalb der Masse, Heterogenität im T1, Tumornekrose, Invasion von Knochen oder Gefäß-Nerven-Strukturen, sowie periphere, knotige, heterogene Kontrastmittelanreicherung sprechen dafür, dass die Masse mit hoher Wahrscheinlichkeit bösartig ist.
Durch den Einsatz von Kontrastmittel im MRT (Kontrastmittel-MRT) lassen sich Hämatome, die Unterscheidung zwischen zystisch und solide, sowie zystisch-nekrotische Anteile in der Masse bestimmen – und so die geeignetste Stelle für die Biopsie auswählen. Allerdings können sowohl zystische Raumforderungen als auch Tumoren mit hohem Myxoidanteil in flüssigkeitssensitiven Sequenzen aufleuchten. Diese lassen sich nur mit Kontrastmittel-MRT zuverlässig unterscheiden. Bei zystischen Raumforderungen verbleibt das Kontrastmittel an der Peripherie, während es bei soliden Massen gleichmäßig im Gewebe aufgenommen wird.
Wenn vor der Operation eine Strahlen- und/oder Chemotherapie durchgeführt wurde, sollte vor dem Eingriff ein neues MRT erfolgen, um das Ansprechen der Behandlung zu bewerten. In manchen Fällen kann die Masse trotz Therapie weiter wachsen – eine mögliche Ursache dafür ist, dass die Masse von innen heraus abstirbt und blutet, was durch eine Kontrastmittel-MRT sichtbar gemacht werden kann.
Sarkome
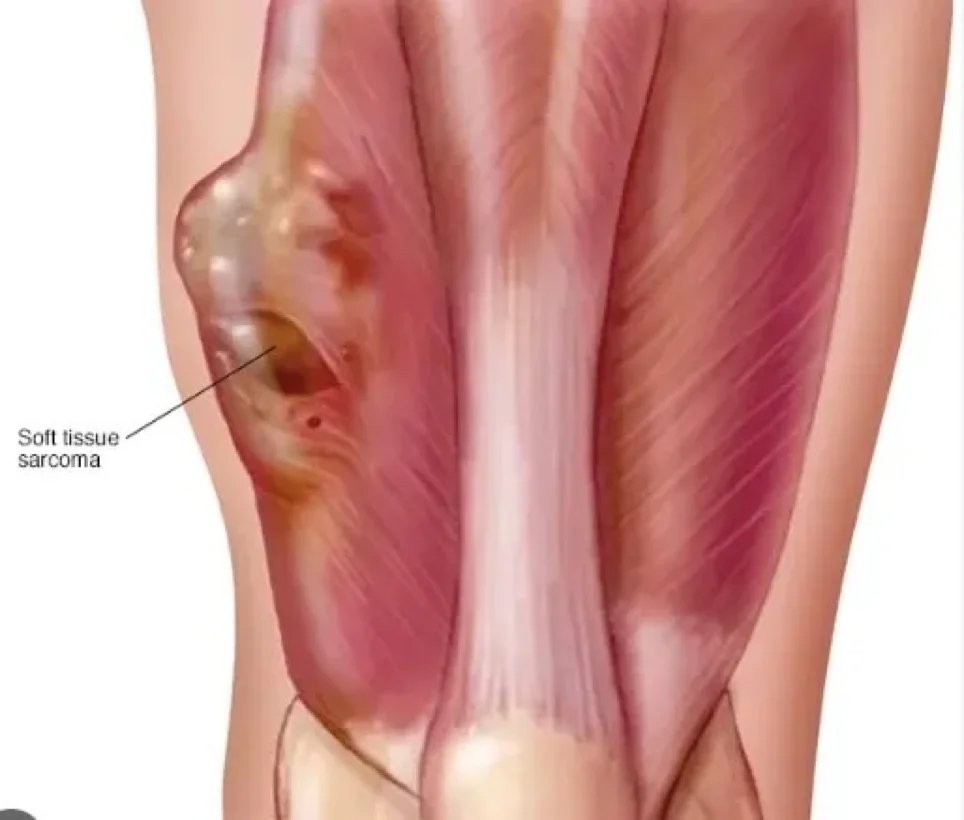
Ein Sarkom ist ein bösartiger Tumor – eine Krebsart, die aus entarteten Zellen mesenchymalen (bindegeweblichen) Ursprungs entsteht. Bindegewebe ist ein Sammelbegriff, der Knochen, Knorpel, Fettgewebe, Gefäß- oder blutbildendes Gewebe umfasst. Sarkome können in all diesen Gewebetypen auftreten. Daher gibt es viele verschiedene Untertypen von Sarkomen, die entsprechend dem spezifischen Ursprungsgewebe und Zelltyp klassifiziert werden. Sarkome sind primäre Bindegewebstumoren, das heißt, sie entstehen direkt aus dem Bindegewebe.
Dies steht im Gegensatz zu sekundären (oder „metastatischen“) Bindegewebstumoren, die entstehen, wenn sich ein Krebs aus einem anderen Teil des Körpers (z. B. Lunge, Brustgewebe oder Prostata) auf das Bindegewebe ausbreitet. Das Wort „Sarkom“ stammt aus dem Griechischen σάρκωμα (sarōma) und bedeutet „fleischige Masse oder Substanz“, abgeleitet von σάρξ (sarx), was „Fleisch“ bedeutet.
Klassifikation
Sarkome werden typischerweise in zwei Hauptgruppen unterteilt: Knochensarkome und Weichteilsarkome, wobei jede Gruppe wiederum mehrere Subtypen umfasst. In den Vereinigten Staaten veröffentlicht das American Joint Committee on Cancer (AJCC) Richtlinien zur Klassifikation der Sarkom-Untergruppen.
Diese Subtypen sind wie folgt:
Subtypen der Knochensarkome:
- Osteosarkom / Chondrosarkom
- Schlecht differenzierte Rund-/Spindelzelltumoren (einschließlich Ewing-Sarkom), Hämangioendotheliom
- Angiosarkom
- Fibrosarkom / Myofibrosarkom / Chordom
- Adamantinom
Weitere:
- Liposarkom / Leiomyosarkom
- Bösartiger peripherer Nervenscheidentumor
- Rhabdomyosarkom
- Synovialsarkom
- Bösartiger solitärer fibröser Tumor
Subtypen der Weichteilsarkome:
- Liposarkom (einschließlich folgender Typen: atypischer lipomatöser Tumor / gut differenziertes Liposarkom, dedifferenziertes Liposarkom, myxoides Liposarkom, pleomorphes Liposarkom und myxoides-pleomorphes Liposarkom)
- Dermatofibrosarcoma protuberans (einschließlich pigmentierter Varianten)
- Dermatofibrosarcoma protuberans, Fibrosarkomatose
- Riesenzell-Fibroblastom
- Bösartiger solitärer fibröser Tumor
- Entzündlicher myofibroblastischer Tumor
- Niedriggradiges myofibroblastisches Sarkom
- Fibrosarkom (einschließlich adulte und sklerosierende epitheloide Varianten)
- Myxofibrosarkom (ehemals myxoides malignes fibröses Histiozytom)
- Niedriggradiges fibromyxoides Sarkom
- Riesenzelltumor des Weichgewebes (Leiomyosarkom)
- Bösartiger Glomustumor
- Rhabdomyosarkom (einschließlich: embryonales, alveoläres, pleomorphes und spindelzelliges/sklerosierendes)
- Hämangioendotheliom (einschließlich: retiformes, pseudomyogenes und epitheloides)
- Weichteilangiosarkom
- Extraskelettales Osteosarkom
- Maligner gastrointestinaler Stromatumor (GIST) / Bösartiger peripherer Nervenscheidentumor (einschließlich epitheloider Variante)
- Maligner Triton-Tumor
- Maligner granularzelliger Tumor
- Maligner ossifizierender fibromyxoidaler Tumor
- Stromales Sarkom, nicht näher bezeichnet
- Myoepitheliales Karzinom
- Maligner phosphaturischer mesenchymaler Tumor
- Synovialsarkom (einschließlich: spindelzellig, biphasisch und nicht näher bezeichnet)
- Epitheloides Sarkom
- Alveoläres Weichteilsarkom
- Klarzelliges Sarkom des Weichgewebes
- Extraskelettales myxoides Chondrosarkom
- Ewing-Sarkom / Interdigitiertes dendritisches Zell-Sarkom
- Desmoplastischer kleinzelliger Rundzelltumor
- Extrarenaler Rhabdoid-Tumor
- Perivaskulärer epitheloider Zelltumor, nicht näher bezeichnet
- Intimales Sarkom
- Undifferenziertes spindelzelliges Sarkom
- Undifferenziertes pleomorphes Sarkom
- Undifferenziertes Rundzellsarkom
- Undifferenziertes epitheloides Sarkom
- Undifferenziertes Sarkom, nicht näher bezeichnet
Sarkom-Symptome:
Zu den typischen Symptomen von Knochensarkomen gehören Knochenschmerzen, insbesondere nachts, sowie Schwellungen im Bereich des Tumors.
Die Symptome von Weichteilsarkomen variieren, treten jedoch meist als harte, schmerzlose Knoten oder Schwellungen auf. Gastrointestinale Stromatumoren (eine Untergruppe der Weichteilsarkome) verlaufen in der Regel asymptomatisch, können jedoch mit unspezifischen Beschwerden wie Bauchschmerzen, Völlegefühl oder anderen Anzeichen eines Darmverschlusses einhergehen.
Ursachen und Risikofaktoren:
Die Ursache der meisten Knochensarkome ist unbekannt, jedoch sind mehrere Faktoren mit einem erhöhten Risiko für die Entwicklung eines Knochensarkoms verbunden. Eine frühere Exposition gegenüber ionisierender Strahlung (z. B. im Rahmen einer Strahlentherapie) ist ein solcher Risikofaktor. Auch die Exposition gegenüber alkylierenden Substanzen, wie sie in bestimmten Chemotherapeutika enthalten sind, erhöht das Risiko. Bestimmte erbliche genetische Syndrome wie das Li-Fraumeni-Syndrom, Mutationen im RB1-Gen und die Paget-Krankheit des Knochens stehen ebenfalls im Zusammenhang mit einem erhöhten Risiko für Knochensarkome.
Die meisten Weichteilsarkome entstehen durch sogenannte „sporadische“ (also zufällige) genetische Mutationen in den betroffenen Zellen. Es gibt jedoch auch bekannte Risikofaktoren: Eine frühere Exposition gegenüber ionisierender Strahlung ist auch hier ein relevanter Risikofaktor. Der Kontakt mit Vinylchlorid (z. B. bei der Herstellung von Polyvinylchlorid – PVC), Arsen und Thorotrast ist mit einem erhöhten Risiko für Angiosarkome verbunden. Auch Lymphödeme, wie sie beispielsweise nach bestimmten Brustkrebsbehandlungen auftreten, können das Risiko für ein Angiosarkom erhöhen. Wie bei Knochensarkomen erhöhen auch hier bestimmte erbliche Syndrome wie das Li-Fraumeni-Syndrom, familiäre adenomatöse Polyposis, Neurofibromatose Typ 1 und RB1-Gen-Mutationen das Risiko für Weichteilsarkome. Das Kaposi-Sarkom wird durch das humane Herpesvirus 8 (HHV-8) verursacht.
Mechanismen:
Obwohl die genauen molekularen Veränderungen, die zur Entstehung von Sarkomen führen, nicht immer bekannt sind, sind einige Sarkomtypen mit bestimmten genetischen Mutationen assoziiert.
Beispiele:
Die meisten Fälle von Ewing-Sarkomen sind mit einer chromosomalen Translokation verbunden, bei der ein Teil von Chromosom 11 mit einem Teil von Chromosom 22 verschmilzt. Dies führt zur Fusion des EWS-Gens mit anderen Genen, darunter in 90 % der Fälle mit dem FLI1-Gen und in 5–10 % der Fälle mit dem ERG-Gen. Diese Genfusionen verursachen die Produktion abnormer Proteine, aber es ist nicht genau bekannt, wie diese Proteine Krebs auslösen.
Das Dermatofibrosarcoma protuberans ist häufig mit einer chromosomalen Translokation verbunden, bei der das COL1A1-Gen mit dem PDGFRB-Gen fusioniert ist. Dies führt zu einer überaktiven PDGF-Signalübertragung, die vermutlich die Zellteilung fördert und letztlich zur Tumorbildung beiträgt.
Der entzündliche myofibroblastische Tumor ist häufig mit Umlagerungen des ALK-Gens und manchmal auch mit Umlagerungen des HMGA2-Gens verbunden.
Der Riesenzelltumor des Weichgewebes ist oft mit einer Translokation zwischen Chromosom 1 und Chromosom 2 verbunden, bei der das CSF1-Gen mit dem COL6A3-Gen fusioniert. Dies führt zu einer vermehrten Produktion des CSF1-Proteins, dem eine Rolle bei der Krebsentwicklung zugeschrieben wird.
Viele Liposarkome sind mit einer Duplikation eines Abschnitts von Chromosom 12 verbunden, was zu zusätzlichen Kopien bekannter krebsfördernder Gene („Onkogene“) wie dem CDK4-Gen, dem MDM2-Gen und dem HMGA2-Gen führt.
Diagnose:
Knochensarkome
Die Diagnose von Knochensarkomen beginnt mit einer ausführlichen Anamnese und körperlichen Untersuchung, die charakteristische Anzeichen und Symptome aufzeigen kann (siehe Anzeichen und Symptome oben). Obwohl einige Knochensarkome (wie Osteosarkom) mit erhöhten alkalischen Phosphatasewerten und andere (wie Ewing-Sarkom) mit einer erhöhten Blutsenkungsgeschwindigkeit einhergehen können, sind Laboruntersuchungen für die Diagnose nicht besonders nützlich. Wichtig ist jedoch, dass keiner dieser Laborbefunde spezifisch für Knochensarkome ist; das bedeutet, dass erhöhte Laborwerte auch mit vielen anderen Erkrankungen außer Sarkomen in Verbindung stehen und daher nicht zur definitiven Diagnose eines Sarkoms herangezogen werden können. Bildgebende Verfahren sind für die Diagnose entscheidend, und die meisten Kliniker veranlassen zunächst eine einfache Röntgenaufnahme. Weitere häufig verwendete bildgebende Verfahren zur Diagnose sind Magnetresonanztomographie (MRT) und radioaktive Knochenszintigrafie. Obwohl sie ein wichtiges Instrument für das Staging ist, wird die Computertomographie (CT) in der Regel nicht zur Diagnose der meisten Knochensarkomarten verwendet. Eine endgültige Diagnose erfordert eine Biopsie des Tumors und eine sorgfältige Untersuchung der Biopsieprobe durch einen erfahrenen Pathologen.
Weichteilsarkome
Die Diagnose von Weichteilsarkomen beginnt mit einer umfassenden Anamnese und körperlichen Untersuchung. Bildgebende Verfahren können CT oder MRT umfassen, wobei CT bei Weichteilsarkomen im Thorax, Abdomen oder Retroperitoneum bevorzugt wird. Die Positronen-Emissions-Tomographie (PET) kann ebenfalls bei der Diagnose hilfreich sein, wird jedoch am häufigsten für das Staging verwendet. Wie bei Knochensarkomen erfordert auch hier die endgültige Diagnose eine Biopsie und die Auswertung der Histologie durch einen geschulten Pathologen.
Staging:
Im Allgemeinen bezieht sich das Krebsstaging darauf, wie weit eine Krebserkrankung fortgeschritten ist, und basiert häufig auf Faktoren wie der Größe des Tumors und der Frage, ob er sich auf andere Körperteile ausgebreitet hat. Das Staging ist wichtig, da das Stadium die Prognose (wahrscheinlicher Verlauf) sowie die Arten von Behandlungen beeinflusst, die wahrscheinlich gegen den Krebs wirksam sind. Beim Staging von Sarkomen wird geprüft, ob der Tumor in umliegendes Gewebe eingedrungen ist (lokale Invasion), sich auf Lymphknoten ausgebreitet hat (nodale Metastasen) oder in andere Gewebe oder Organe im Körper eingedrungen ist (Fernmetastasen).
Die am häufigsten verwendeten bildgebenden Verfahren zur Stadieneinteilung von Knochensarkomen sind MRT oder CT zur Beurteilung des Primärtumors, kontrastverstärkte CT des Brustkorbs zur Beurteilung, ob sich der Krebs auf die Lunge ausgebreitet hat (d. h. metastasiert ist), und eine radioaktive Knochenszintigrafie zur Beurteilung von Knochenmetastasen. Das Staging von Weichteilsarkomen umfasst typischerweise die Bildgebung des Primärtumors mittels MRT oder CT zur Bestimmung der Tumorgröße sowie eine kontrastverstärkte CT des Brustkorbs zur Beurteilung metastatischer Tumoren in der Lunge.
Grad:
Wie bei einigen anderen Krebsarten wird auch bei Sarkomen ein Grad (niedrig, mittel oder hoch) anhand des mikroskopischen Erscheinungsbilds der Tumorzellen eingeteilt. Der Grad gibt im Allgemeinen an, wie aggressiv der Krebs ist und wie wahrscheinlich eine Ausbreitung („Metastasierung“) in andere Körperregionen ist. Niedriggradige Sarkome haben eine bessere Prognose als hochgradige Sarkome und werden in der Regel operativ behandelt. Mittel- und hochgradige Sarkome werden häufiger mit einer Kombination aus Operation, Chemotherapie oder Strahlentherapie behandelt. Da höhergradige Tumoren häufiger metastasieren (invasiv und sich in regionale und entfernte Regionen ausbreiten), werden sie aggressiver behandelt. Das Wissen, dass viele Sarkome empfindlich auf Chemotherapie reagieren, hat die Überlebenschancen der Patienten deutlich verbessert. Beispielsweise lag die Langzeitüberlebensrate bei pädiatrischen Patienten mit lokalisiertem Osteosarkom vor der Chemotherapie bei nur 20 %, heute ist sie auf 60–70 % gestiegen.
Behandlung
Die häufigste Behandlungsmethode für die meisten Sarkome, die sich nicht auf andere Körperteile ausgebreitet haben, ist die Operation. Wir führen heute eine gliedmaßenerhaltende Operation anstelle einer Amputation durch, um die Gliedmaßen der Patienten in mindestens 90 % der Fälle von Extremitätensarkomen (Arm oder Bein) zu erhalten. Zusätzliche Behandlungen, darunter Chemotherapie, Strahlentherapie (auch „Radiotherapie“ genannt) und Protonentherapie, können vor der Operation (sogenannte „neoadjuvante“ Chemotherapie oder Radiotherapie) oder nach der Operation (sogenannte „adjuvante“ Chemotherapie oder Radiotherapie) durchgeführt werden. Der Einsatz der neoadjuvanten oder adjuvanten Chemotherapie und Radiotherapie hat die Prognose vieler Sarkompatienten deutlich verbessert. Die Behandlung kann ein langer und mühsamer Prozess sein und bei vielen Patienten etwa ein Jahr dauern.
Prognose
Prognosebeeinflussende Faktoren
Das AJCC hat mehrere Faktoren identifiziert, die die Prognose von Knochensarkomen beeinflussen:
Tumorgröße: Größere Tumoren haben tendenziell eine schlechtere Prognose als kleinere Tumoren.
Tumorausbreitung in umliegendes Gewebe: Tumoren, die sich lokal in umliegendes Gewebe ausgebreitet haben, haben tendenziell eine schlechtere Prognose als Tumoren, die sich nicht über ihren Ursprungsort hinaus ausgebreitet haben.
Stadium und Vorhandensein von Metastasen: Tumoren, die sich in die Lymphknoten (selten bei Knochensarkomen) oder andere Organe oder Gewebe (z. B. die Lunge) ausgebreitet haben, haben eine schlechtere Prognose als Tumoren, die sich nicht ausgebreitet haben.
Tumorgrad: Hochgradige Tumoren (Grad 2 und 3) haben tendenziell eine schlechtere Prognose als niedriggradige (Grad 1).
Skelettale Lokalisation: Tumoren, die von der Wirbelsäule oder den Beckenknochen ausgehen, haben tendenziell eine schlechtere Prognose als Tumoren, die von den Knochen der Arme oder Beine ausgehen.
Faktoren, die die Prognose für Weichteilsarkome (mit Ausnahme von GIST) beeinflussen, sind:
Grad: Die AJCC empfiehlt die Verwendung eines Graduierungssystems namens „French Federation of Cancer Centers Sarcoma Group (FNCLCC) Grade“ für Weichteilsarkome, bei dem höhergradige Tumoren eine schlechtere Prognose haben als niedriggradige Tumoren. p>
Wichtiger Faktor für die Prognose von GISTs:
Mitoserate: Die Mitoserate bezeichnet den Anteil der Zellen, die sich im Tumor aktiv teilen. GISTs mit einer hohen Mitoserate haben eine schlechtere Prognose als GISTs mit einer niedrigen Mitoserate.
Ergebnisdaten
Laut Daten des US-amerikanischen National Cancer Institute (NCI) beträgt die 5-Jahres-Gesamtüberlebensrate bei Knochensarkomen 66,9 %. Die American Cancer Society (ACS) schätzt, dass im Jahr 2019 in den USA 1.660 Menschen an Knochensarkomen sterben werden, was 0,3 % aller Krebstodesfälle entspricht. Das durchschnittliche Sterbealter liegt bei 61 Jahren, der Tod kann jedoch in jeder Altersgruppe eintreten. So ereignen sich 12,3 % der Todesfälle durch Knochensarkom bei Menschen unter 20 Jahren, 13,8 % bei Menschen zwischen 20 und 34 Jahren, 5,5 % bei Menschen zwischen 35 und 44 Jahren und 9,3 % bei Menschen zwischen 45 und 54 Jahren. 13,5 % bei Personen im Alter von 55–64 Jahren, 16,2 % bei Personen im Alter von 65–74 Jahren, 16,4 % bei Personen im Alter von 75–84 Jahren und 13,1 % bei Personen ab 85 Jahren. Es tritt bei Personen über 10 Jahren auf.
Die 5-Jahres-Gesamtüberlebensrate bei Weichteilsarkomen (unabhängig vom Stadium) beträgt 64,5 %, wobei das Überleben von vielen Faktoren, einschließlich des Stadiums, beeinflusst wird. Daher beträgt die 5-Jahres-Überlebensrate 80,8 % bei Weichteilsarkomen, die sich nicht über den Primärtumor hinaus ausgebreitet haben („lokalisierte“ Tumoren), 58,0 % bei Weichteilsarkomen, die sich nur auf nahegelegene Lymphknoten ausgebreitet haben, und 16 % bei Weichteilsarkomen mit Fernorganausbreitung. Die ACS schätzt, dass im Jahr 2019 5.270 Menschen an Weichteilsarkomen sterben werden, was 0,9 % aller Krebstodesfälle entspricht.
Epidemiologie
Sarkome sind extrem selten. Das Risiko, dass bei einer zuvor gesunden Person erneut Knochenkrebs diagnostiziert wird, liegt unter 0,001 % (1 von 100.000), während das Risiko, an einem Weichteilsarkom zu erkranken, zwischen 0,0014 und 0,005 % liegt. Die American Cancer Society schätzt, dass es in den USA im Jahr 2019 3.500 neue Fälle von Knochensarkomen und 12.750 neue Fälle von Weichteilsarkomen geben wird. Bei einer geschätzten Gesamtzahl von 1.762.450 Krebsneudiagnosen (alle Krebsarten) bedeutet dies, dass Knochensarkome nur 0,2 % aller Krebsneudiagnosen ausmachen (und damit die 30. häufigste Krebsart). Weichteilsarkome machten 2019 in den USA nur 0,7 % aller Krebsneudiagnosen aus (und sind damit die 22. häufigste Krebsart).
Sarkome betreffen Menschen jeden Alters. Etwa 50 % der Knochensarkome und 20 % der Weichteilsarkome werden bei Menschen unter 35 Jahren diagnostiziert. Einige Sarkome, wie Leiomyosarkom, Chondrosarkom und gastrointestinaler Stromatumor (GIST), treten bei Erwachsenen häufiger auf als bei Kindern. Die meisten hochgradigen Knochensarkome, darunter Ewing-Sarkom und Osteosarkom, kommen bei Kindern und jungen Erwachsenen deutlich häufiger vor.
Behandlung:
Die Behandlung von Sarkomen erfordert häufig eine Chemotherapie, insbesondere wenn das Sarkom bereits gestreut oder metastasiert ist. Aktuelle Chemotherapeutika sind jedoch mit erheblichen Toxizitäten verbunden und können Krebszellen nicht vollständig abtöten. Daher wird seit 2019 an der Entwicklung neuer Medikamente zur Behandlung von Sarkomen geforscht. Eine Möglichkeit ist die Krebsimmuntherapie (z. B. Immun-Checkpoint-Inhibitoren wie Anti-PD1-, Anti-PDL1- und Anti-CTLA4-Wirkstoffe) zur Behandlung von Sarkomen. Dies ist jedoch noch kein etabliertes Behandlungsinstrument. Weitere Strategien wie die zielgerichtete Therapie mit niedermolekularen Substanzen, biologische Wirkstoffe (z. B. kleine interferierende RNA-Moleküle) und die nanopartikelfokussierte Therapie werden ebenfalls untersucht.
Dr. Sameer Rastogi et al. hat bei mehreren Sarkomen (UPS & ASPS) lang anhaltende Reaktionen auf Immuntherapien gezeigt.
Die Forschung erforscht weiterhin die spezifischen genetischen und molekularen Faktoren, die zur Entstehung von Sarkomen führen. Dies könnte die Entwicklung neuer zielgerichteter Therapien ermöglichen und es Ärzten ermöglichen, die Prognose eines Patienten genauer vorherzusagen.
Das Vorhandensein des immunregulatorischen Checkpoint-Rezeptors H3-B3 auf Tumorzellen bietet die Möglichkeit für klinische Studien zur Erprobung neuer Medikamente, zielgerichteter Wirkstoffe und Immuntherapien in der Entwicklung.
Bewusstsein:
In den USA gilt der Juli als Sarkom-Bewusstseinsmonat. In Großbritannien findet im Juli die Sarkom-Bewusstseinswoche statt, die von der Wohltätigkeitsorganisation Sarcoma UK für Knochen- und Weichteilkrebs organisiert wird.
Der amerikanische YouTuber Technoblade erhielt im August 2021 die Diagnose Sarkom und verstarb im Juni 2022 an den Folgen seiner Erkrankung, nachdem der Krebs Metastasen gebildet hatte. Er sammelte über 500.000 US-Dollar in einer Wohltätigkeitskampagne. Viele YouTuber schärften das Bewusstsein und spendeten an Wohltätigkeitsorganisationen wie die Sarcoma Foundation of America, nachdem Technoblade diagnostiziert worden war und verstarb.
Biopsie
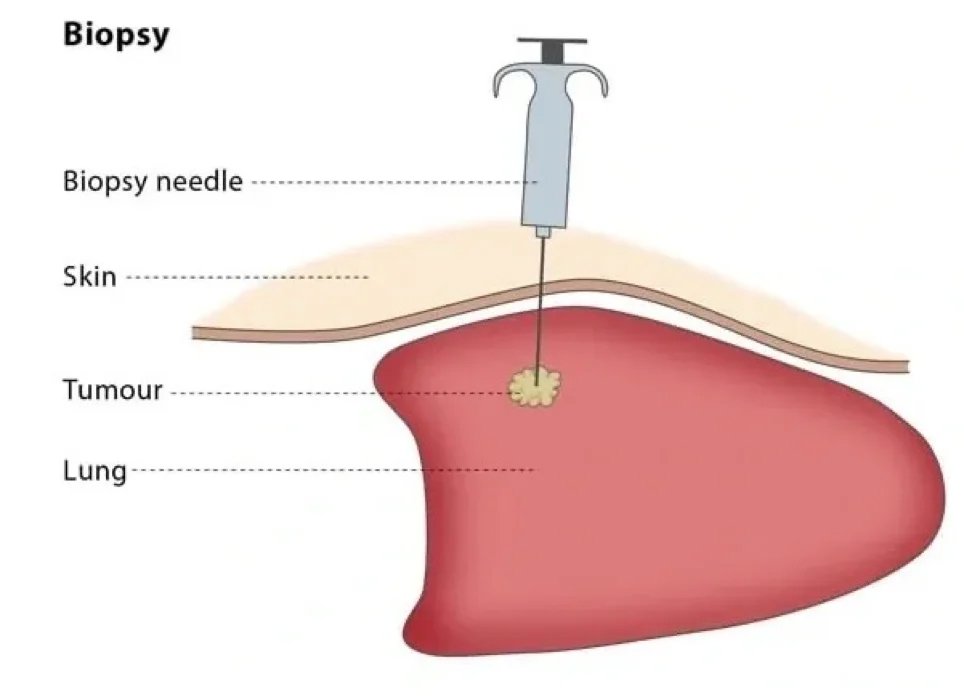
Biopsieprinzipien:
- Eine Krebsoperation kann nicht ohne Krebsdiagnose durchgeführt werden.
- Die klinische und radiologische Untersuchung muss vor der Biopsie abgeschlossen sein.
- Obwohl gutartige Tumoren deutlich häufiger sind als bösartige, sollte die Diagnose Krebs immer in Betracht gezogen werden.
- Eine geschlossene Biopsie ist in den meisten Fällen ausreichend und sollte bevorzugt werden.
- Sollte eine offene Biopsie durchgeführt werden, muss diese vom Chirurgen durchgeführt werden, der die Operation durchführen wird.
- Die sogenannte Exzisionsbiopsie (bei der die gesamte Tumormasse als Biopsiematerial betrachtet wird, ohne Teile zu entnehmen und eine weite Resektion durchzuführen) sollte nur in besonderen Fällen angewendet werden.
- Die Diagnose und Behandlung von Sarkomen ist Teamarbeit. Es ist notwendig, diese Aufgabe in kompetente Hände zu legen.
Wenn diese Grundsätze beachtet werden, kann die genaueste Diagnose mit dem geringsten Schaden für den Patienten gestellt werden. Diese sind:
- Lipome (Fettdrüsen): Ist die Läsion in allen Sequenzen homogen und isointens mit dem Fettgewebe, ist keine Biopsie erforderlich.
- Gutartige Knochentumoren mit typischer Radiologie (Ostechondrome, nicht-ossifizierende Fibrome, Enchondrome, unikamerale einfache Knochenzysten). Diese Läsionen erfordern möglicherweise keine weitere Untersuchung mit hochwertigen Röntgenstrahlen.
- Neben Lipomen (Weichteiltumoren mit typischen MRT-Befunden) sind Schwannome, Hämangiome, arteriovenöse Malformationen und Myxome zu berücksichtigen.
- Metastasierte Läsionen bei Patienten mit bioptisch nachgewiesenem Karzinom oder Myelom.
Weichteilbiopsien führe ich in der Regel unter örtlicher Betäubung in der Praxis durch. Die Biopsie mit einer Nadel dauert etwa zehn Minuten und ist schmerzarm. Der Patient kann noch am selben Tag wieder arbeiten. Obwohl ich mit einem unserer erfahrensten Dozenten für geschlossene Nadelbiopsien zusammenarbeite, kann selten eine Diagnose gestellt werden. Einige Autoren empfehlen daher die Umstellung auf eine offene Biopsie, während andere die Fortsetzung der geschlossenen Biopsie bis zur Diagnose empfehlen. Wenn ich zweimal eine geschlossene Biopsie versuche und immer noch keine Diagnose stelle, ziehe ich es vor, auf eine offene Biopsie umzusteigen.
Knochenbiopsien führe ich im Operationssaal unter bildgebender Szintigraphie durch, vorzugsweise als geschlossene Nadelbiopsie. Ich entnehme die Biopsieproben unter Vollnarkose. Der Eingriff dauert etwa 15 Minuten, sodass der Patient keine Schmerzen verspürt.
Wenn bei Bildgebung, Blutuntersuchungen oder einer Untersuchung vor dem Eingriff der Verdacht auf eine Infektion besteht, sollten die Antibiotika abgesetzt, mindestens zwei Wochen gewartet und Kulturen auf aerobe, anaerobe, säurefeste Bakterien und Pilze angelegt werden. Früher war es üblich, von jeder Biopsie eine Kultur und von jeder Kultur eine Biopsie zu entnehmen. Dank moderner Bildgebungs- und Labormethoden ist es jedoch möglich, das Infektionsrisiko bei Biopsiepatienten einzugrenzen. Unnötige Kulturen können zu einer Fehldiagnose einer Infektion führen, was den Patienten langen, anstrengenden und teuren Antibiotikabehandlungen aussetzen kann.

Osteosarcoma patient
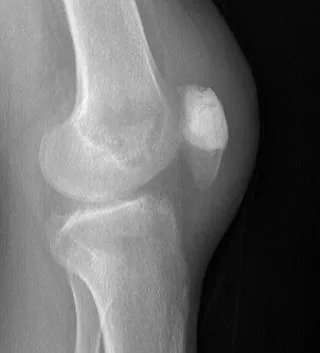
Patella Aneurysmal Bone Cyst
Extremely rare case
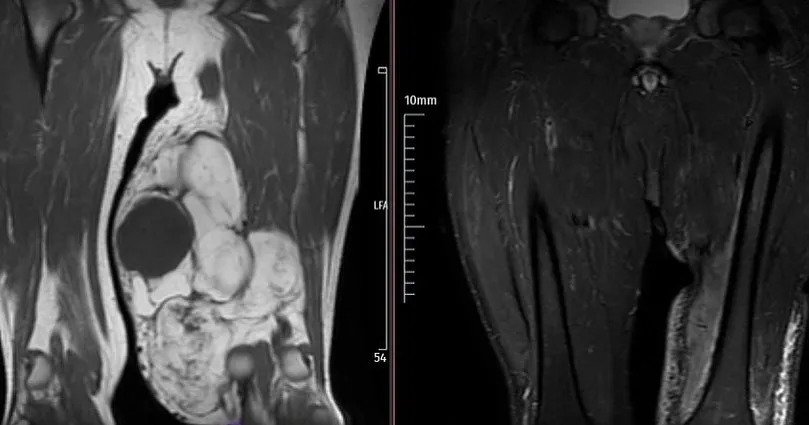
Dedifferentiated Liposarcoma Case
No recurrence with curettage and cementation
Dedifferentiated Chondrosarcoma
The residual tumor in the patient, who underwent inadequate surgery 10 years ago, had deteriorated and reached a gigantic size. Extensive resection was performed and the patient regained his health.

Giant Cell Bone Tumor
It is very rare and very malignant but we have not had any recurrence or metastasis for two years.
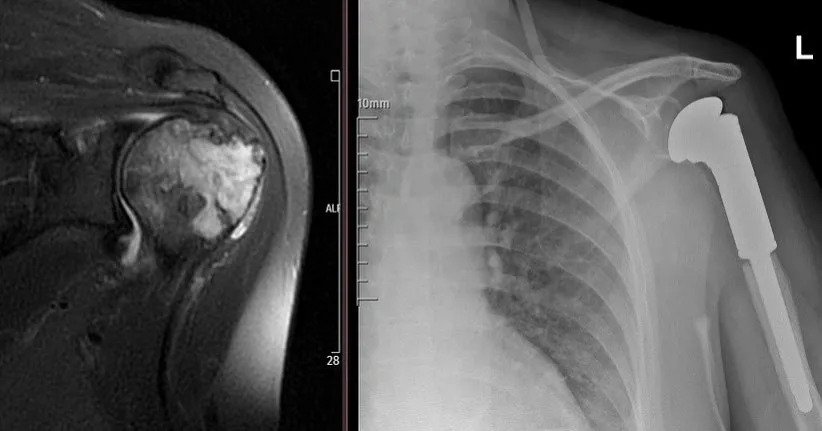
Soft tissue Ewing Sarcoma
Giant cell tumor is benign but has a devastating effect on bone
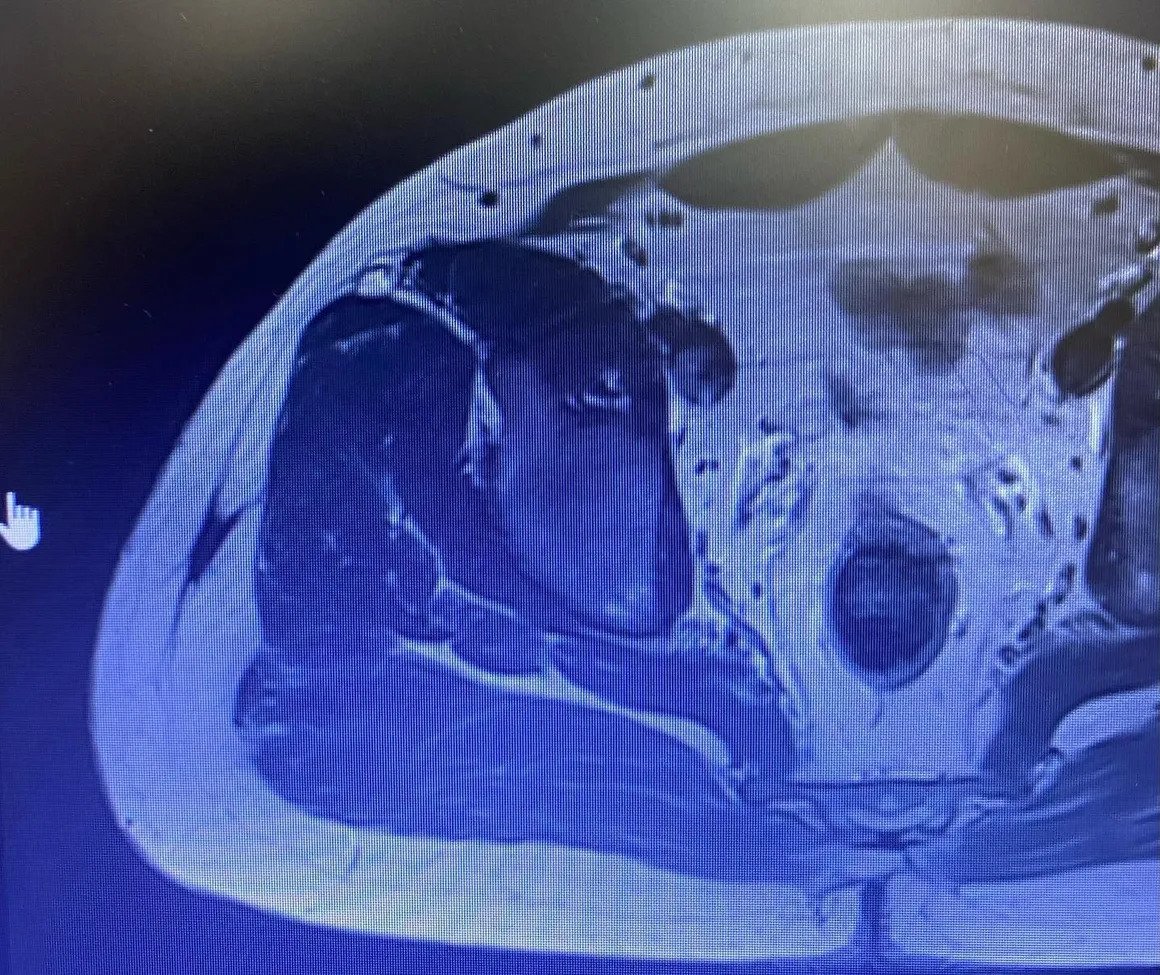
Soft tissue Ewing Sarcoma
Soft tissue Ewing sarcomas are extremely rare. The patient regained his health with chemotherapy and surgery
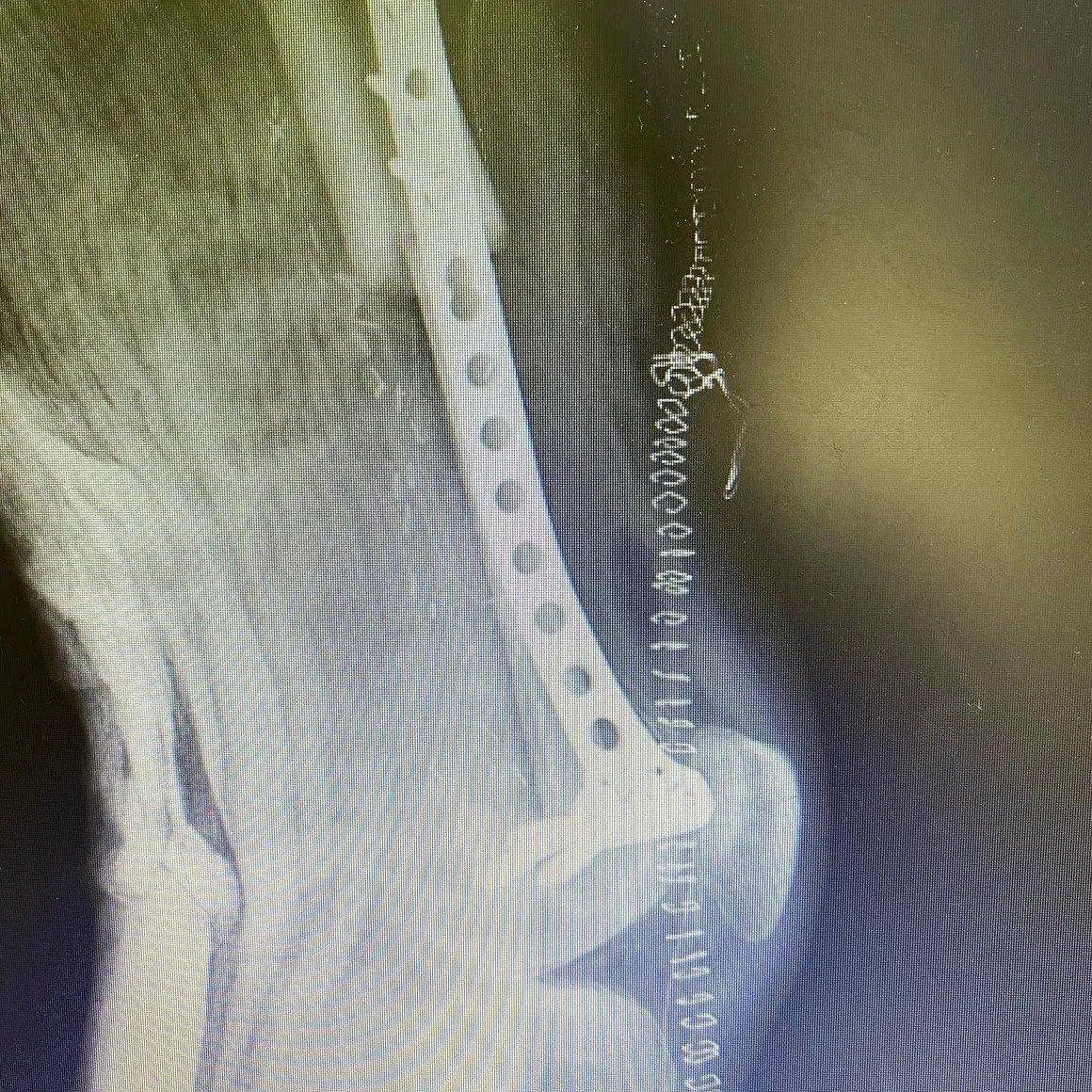
Femur Lower End Osteosarcoma
Soft tissue Ewing sarcomas are extremely rare. The patient regained his health with chemotherapy and surgery
Femur Lower End Osteosarcoma
After chemotherapy, extensive resection and vascularized fibula bone transplantation saved his leg.

Femur Lower End Osteosarcoma

Atypical Cartilage Tumor
After chemotherapy, extensive resection and vascularized fibula bone transplantation saved his leg.

Osteoid Osteoma Case
It's not cancer, but it can turn into cancer. He got rid of the mass completely with open surgery.
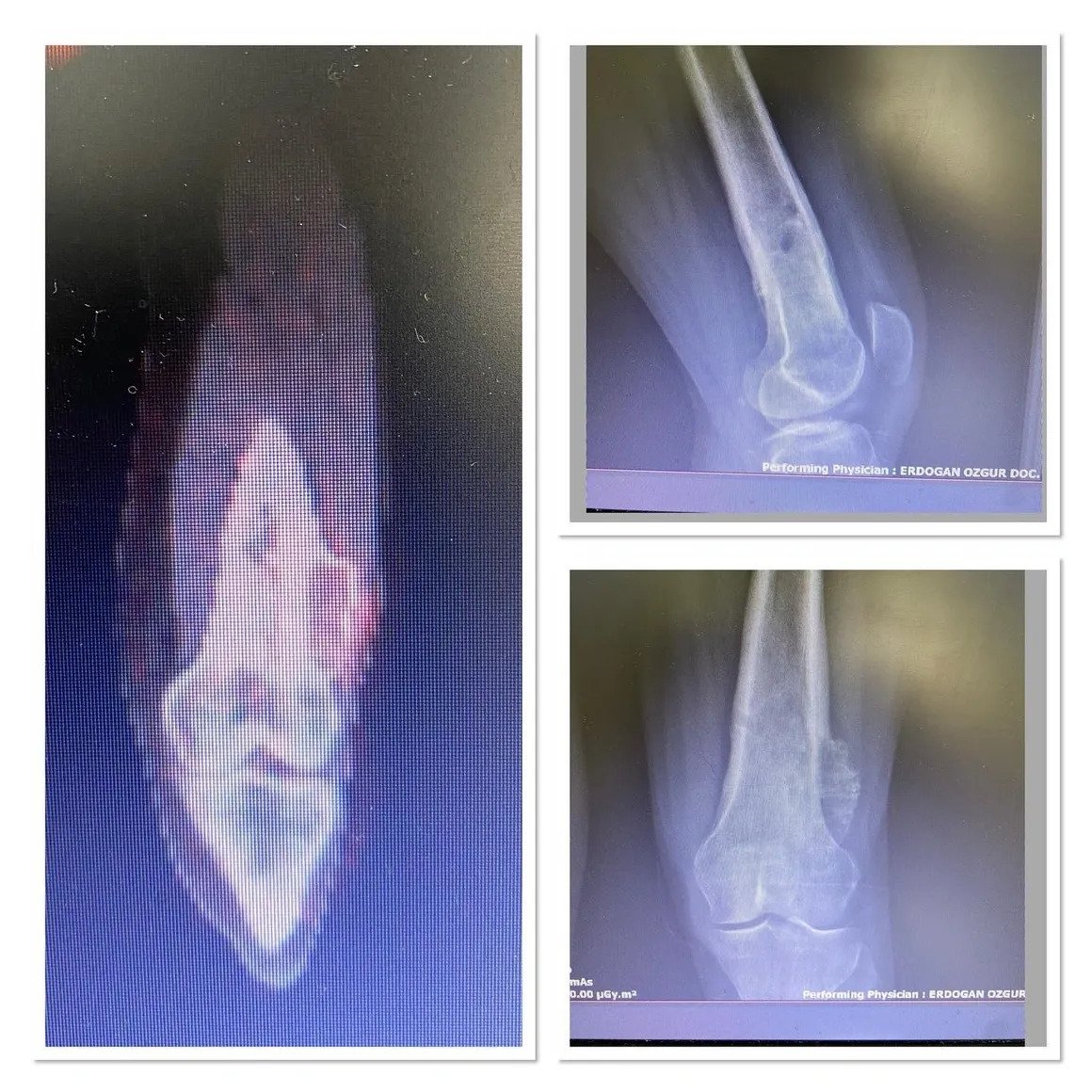
Fibula Osteosarcoma
With curettage, the pain disappeared the next day

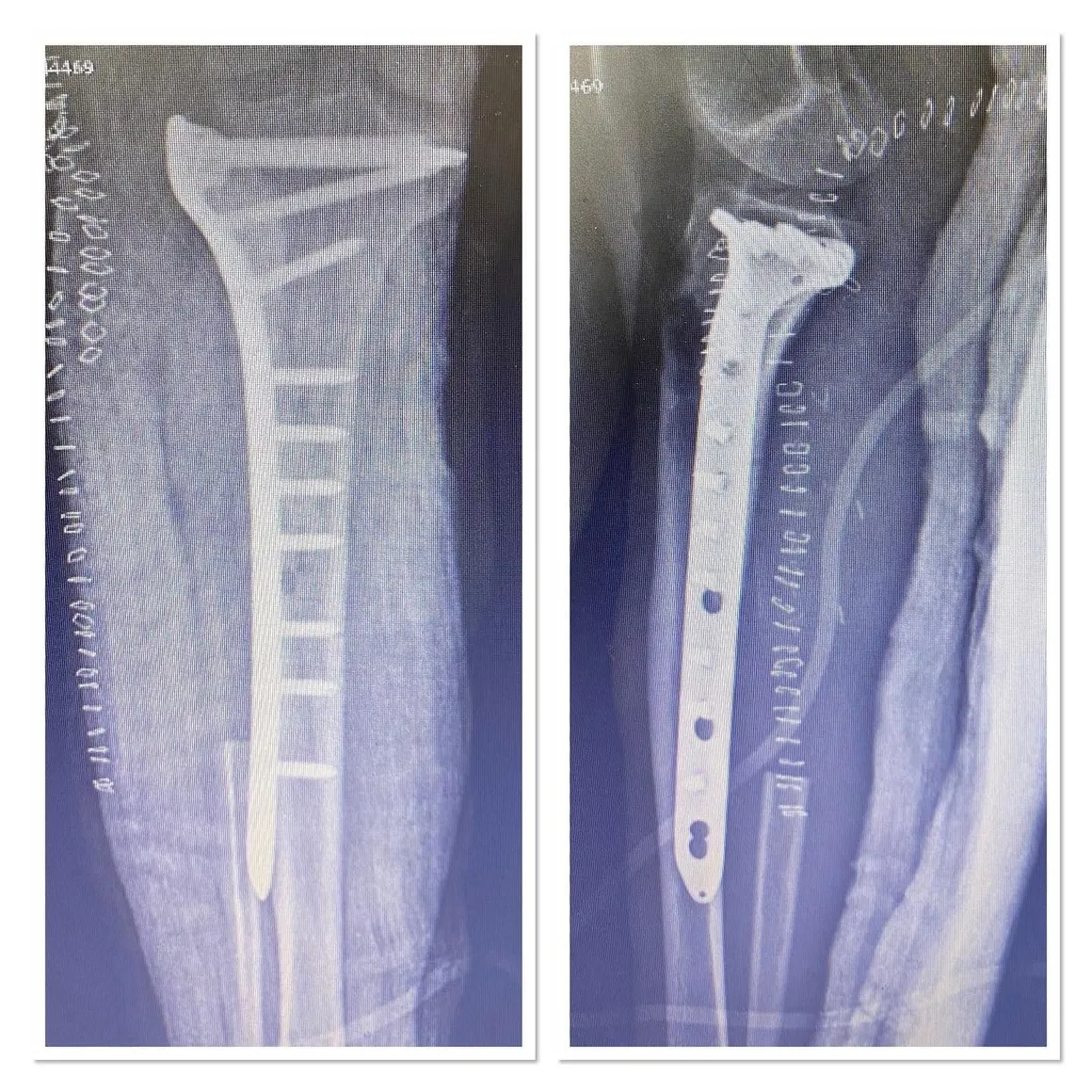
Fibula Osteosarcoma
Fibula tumors are one of the most difficult orthopedic tumor surgeries after pelvic tumors. In this patient, the tumor was extensively removed and vascular repair was performed.

Fibula Osteosarcoma
Fibula tumors are one of the most difficult orthopedic tumor surgeries after pelvic tumors. In this patient, the tumor was extensively removed and vascular repair was performed.
