




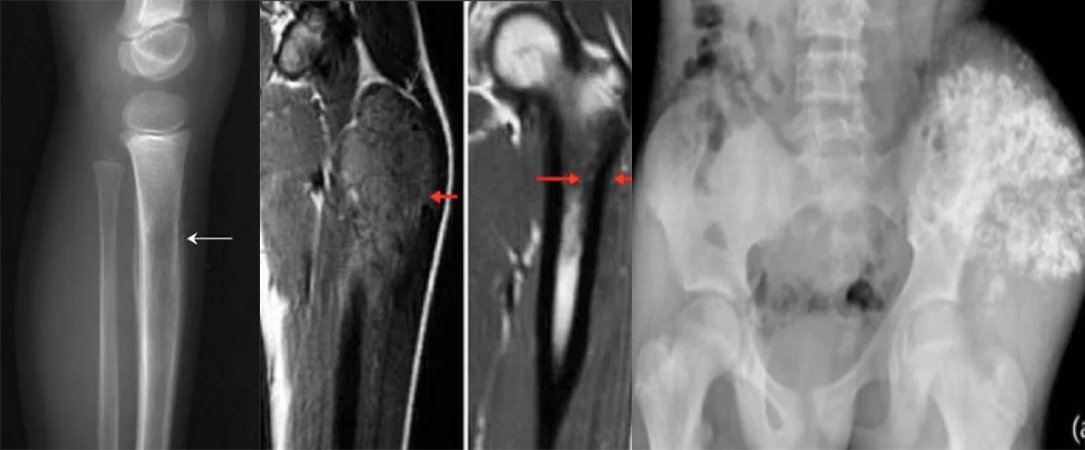
BEHANDLUNG DES EWING-SARKOMS
Das Ewing-Sarkom ist der zweithäufigste primär maligne Knochentumor und betrifft hauptsächlich Jugendliche im zweiten Lebensjahrzehnt. Es handelt sich um eine hochmetastasierende Sarkomform. Trotz Strahlentherapie oder chirurgischer Eingriffe starben in der Vergangenheit schätzungsweise 85 % bis 90 % der Patienten innerhalb weniger Monate nach Metastasierung. Heute jedoch, dank bedeutender Fortschritte in der Behandlung – sowohl durch lokale Therapien als auch durch eine Kombination aus verschiedenen Chemotherapeutika – ist die 5-Jahres-Überlebensrate von unter 20 % auf über 70 % gestiegen. Die Rückfallquote bleibt jedoch weiterhin hoch. Eine frühzeitige Erkennung und Behandlung ist entscheidend, um eine hohe Morbidität und Mortalität zu verhindern. Das Ewing-Sarkom (ES) ist ein aggressiver Tumor, der bei Jugendlichen und jungen Erwachsenen auftritt und etwa 10 % bis 15 % aller Knochensarkome ausmacht.
Das Ewing-Sarkom wurde erstmals 1921 von James Ewing beschrieben. Zur Ewing-Sarkom-Familie gehören das „klassische“ knöcherne Ewing-Sarkom, das extraskelettale Ewing-Sarkom, der maligne kleinzellige Tumor der Brustwand (Askin-Tumor) sowie die aus dem Weichgewebe stammenden primitiven neuroektodermalen Tumoren (PNET). Die Translokation t(11;22)(q24;q12) ist mit 85 % der Tumoren assoziiert und führt zur Bildung des Fusionsgens EWS-FLI-1, während t(21;12)(22;12) und andere, seltener vorkommende Translokationen ebenfalls EWS-FLI-1 oder verwandte ERG-Fusionen verursachen. Letztere machen die restlichen 10 % bis 15 % der Fälle aus. Die häufigsten anatomischen Lokalisationen sind das Becken, das axiale Skelett und der Femur; jedoch kann das Ewing-Sarkom nahezu in jedem Knochen oder Weichgewebe auftreten. Typischerweise äußert es sich durch Schmerzen und Schwellung im betroffenen Bereich. Obwohl die meisten Tumoren zunächst lokal erscheinen, weisen nahezu alle Patienten eine subklinische metastatische Erkrankung auf. Etwa 25 % der Patienten mit zunächst lokalisiertem Tumor erleiden letztlich ein Rezidiv.
Für rezidivierende und refraktäre Ewing-Sarkome (ES) gibt es keine Standardtherapie. Es besteht kein Zusammenhang zwischen ES und Umweltfaktoren, Medikamentenexposition, früherer Strahlenbelastung oder familiärer Krebsvorgeschichte. Das Ewing-Sarkom besteht aus kleinen, runden Zellen mit einem erhöhten Nukleus-Zytoplasma-Verhältnis und gehört zur Familie der kindlichen „small round blue cell tumors“ (z. B. Retinoblastom, Neuroblastom, Rhabdomyosarkom und Nephroblastom). Ewing-Zellen besitzen wenig eosinophiles Zytoplasma mit reichlich Glykogen, welches typischerweise durch Periodic-Acid-Schiff (PAS)-Färbung nachgewiesen wird. Eine hohe CD99-Expression wurde in über 80 % der Fälle nachgewiesen. Dieser hochsensitive immunhistochemische Marker spielt vermutlich eine Rolle bei der Migration von Leukozyten zum Endothel, ist jedoch nicht spezifisch, da er auch in anderen Sarkomen und Lymphomen vorkommen kann. Neben dem MIC2-Genprodukt CD99 exprimieren Ewing-Zellen häufig auch CD45, Synaptophysin, Chromogranin, Vimentin, Keratin, Desmin, neuronenspezifische Enolase (NSE) und S-100. Diese immunhistochemische Diagnostik ist jedoch in ihrer Aussagekraft begrenzt, da sie keine ausreichende Spezifität bietet. Zur definitiven Diagnosestellung sind molekulargenetische Untersuchungen mittels FISH (Fluoreszenz-in-situ-Hybridisierung) und/oder RT-PCR (Reverse-Transkriptions-Polymerase-Kettenreaktion) erforderlich.
Befunde bei Ewing-Sarkom: Patient:innen mit Ewing-Sarkom stellen sich typischerweise mit lokalen Symptomen wie Schmerzen, Steifheit oder Schwellungen vor, die über Wochen oder Monate anhalten. Mehr als 50 % berichten über intermittierende Schmerzen, die nachts zunehmen. Ewing-Sarkome können an vielen verschiedenen Stellen mit unterschiedlichen klinischen Erscheinungen auftreten, meist jedoch in der Diaphyse langer Röhrenknochen. Knochen- oder Metastasenläsionen können sich als pathologische Frakturen manifestieren. Bei Beckenbefall äußert sich die Erkrankung möglicherweise als Rückenschmerz. Systemische Symptome wie Fieber und Gewichtsverlust deuten häufig auf eine metastasierte Erkrankung hin. Etwa 20 % der Patient:innen haben zum Diagnosezeitpunkt bereits Fernmetastasen, bei über 20 % dieser Fälle liegt ein Befall von Lunge oder Pleura vor.
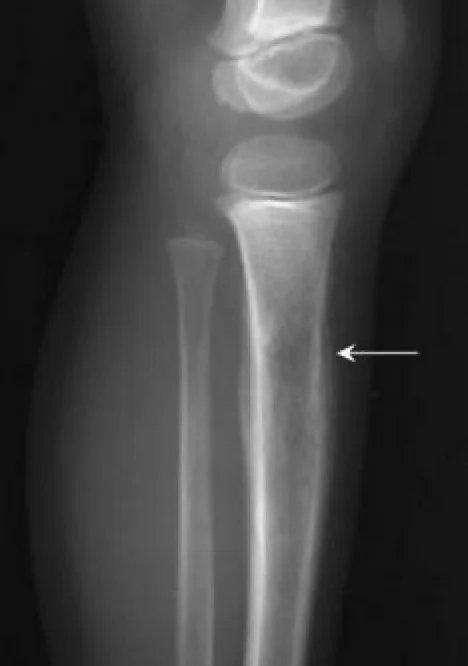
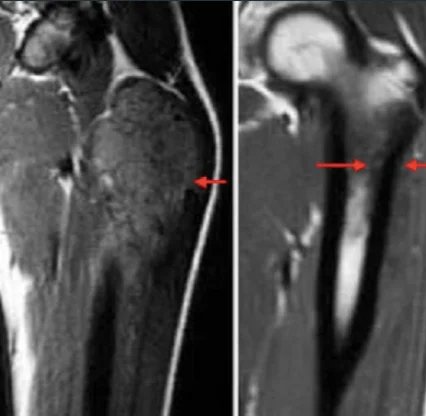
Eine gründliche klinische Untersuchung ist essenziell. Bei Lungen- und Pleurabefall können asymmetrische Atemgeräusche, pleurale Beschwerden oder Rasselgeräusche auffallen. Bei Knochenmarkmetastasen können aufgrund von Thrombozytopenie Petechien oder Purpura auftreten. Bei ZNS-Beteiligung ist auch eine neurologische Untersuchung wichtig. Erste bildgebende Verfahren beinhalten ein Röntgenbild der betroffenen Region; typisch ist hier das sogenannte „Zwiebelschalen“-Muster als periostale Reaktion. Der Primärtumor und mögliche Metastasierungsregionen sollten bildgebend abgeklärt werden. Konventionelle Röntgenaufnahmen können destruierende, mottenfraßartige Läsionen, ein Codman-Dreieck oder multilamelläre periostale Reaktionen („Onion Skin“) zeigen. Laut der aktualisierten Leitlinie des National Comprehensive Cancer Network (NCCN) von 2017 erfolgt die Primärdiagnostik mittels MRT und gegebenenfalls CT, wobei MRT mit Kontrastmittel von zentraler Bedeutung ist. Weitere bildgebende Verfahren umfassen Thorax-CT, PET/CT sowie MRT von Wirbelsäule und Becken. Wenn eine Biopsie notwendig ist, sollte die/der Patient:in an eine:n orthopädische:n Tumorchirurg:in überwiesen werden. Die Diagnose wird bevorzugt mittels Nadelbiopsie oder offener Biopsie gestellt. Eine molekularzytogenetische Untersuchung der Biopsieproben zur Analyse der Translokation t(11;22) sollte Teil der Diagnostik sein. Knochenmarkpunktion und -biopsie können erwogen werden. Gemäß NCCN-Leitlinie sollte die initiale Diagnostik auch eine Bestimmung der Serum-LDH (Laktatdehydrogenase) umfassen, da dieser Wert prognostische Bedeutung hat. Zudem sollte allen Patient:innen eine Fertilitätsberatung (z. B. Kryokonservierung von Spermien oder Eizellen) vor Beginn der Therapie angeboten werden.
Chemotherapie und Strahlentherapie beim Ewing-Sarkom: Historisch verglichen die Intergroup-Ewing-Sarcoma-Studien (IESS-I und IESS-II) die Kombination aus RT+VACA (Vincristin, Dactinomycin und Cyclophosphamid) mit VAC (Vincristin, Dactinomycin und Cyclophosphamid). Die Kombination mit Cyclophosphamid und Doxorubicin zeigte bessere Ergebnisse. Aufgrund der Dosisbegrenzung von Doxorubicin in Dactinomycin-haltigen Protokollen zeigten nachfolgende Studien keinen signifikanten Einfluss auf das klinische Ergebnis, wenn Dactinomycin weggelassen wurde. Mehrere Studien untersuchten die Zugabe von Ifosfamid und Etoposid zur Standardchemotherapie. Die INT0091-Studie der Pediatric Oncology Group – Child Cancer Group zeigte, dass die VACD-IE-Gruppe signifikant bessere Überlebensraten aufwies als die VACD-Gruppe.
Darüber hinaus war die Inzidenz lokaler Rückfälle in der VACD-IE-Gruppe geringer. In der EICESS-92-Studie (European Intergroup Cooperative Ewing Sarcoma Study) wurden bei Patienten mit Standardrisiko (SR) VACA (Vincristin, Dactinomycin, Cyclophosphamid und Doxorubicin) und VAIA (Vincristin, Dactinomycin, Ifosfamid und Doxorubicin) miteinander verglichen. Es zeigte sich, dass die Wirkung von Cyclophosphamid der von Ifosfamid ähnlich war; jedoch wurde Cyclophosphamid mit einer erhöhten Toxizität in Verbindung gebracht. Die 3-Jahres-Ereignisfreie-Überlebensrate (EFS) betrug 73 % bei VACA und 74 % bei VAIA. Die Euro-EWING99-R1-Studie (eine Äquivalenzstudie basierend auf dem EICESS-92-Protokoll) untersuchte, ob Cyclophosphamid Ifosfamid in der Konsolidierungstherapie (einschließlich Vincristin und Dactinomycin) bei Patienten mit Standardrisiko ersetzen könne, und verglich VAC (Vincristin, Dactinomycin und Cyclophosphamid) mit VAI (Vincristin, Dactinomycin und Ifosfamid). Die Ergebnisse deuten darauf hin, dass VAC statistisch nicht unterlegen war; allerdings war die 3-Jahres-EFS unter VAI geringfügig höher. In einer Phase-III-Studie (AEWS0031) der Children's Oncology Group (COG) erhielten die Patienten im Wechsel VDC (Vincristin, Doxorubicin, Cyclophosphamid) und IE (Ifosfamid, Etoposid) alle drei Wochen. Die Studie zeigte, dass 2-wöchige Intervalle wirksamer waren als 3-wöchige Intervalle, ohne die Toxizität zu erhöhen.
Diese Ergebnisse führten dazu, dass VDC/IE in den USA zum Standardbehandlungsprotokoll wurde. Die Chemotherapie beginnt vor der lokalen Behandlung und wird nach der Operation fortgesetzt, sofern keine Krankheitsprogression vorliegt. Die lokale Kontrolle erfolgt durch chirurgische Resektion und/oder Strahlentherapie. Bis heute gibt es keine Studien, die die Wirksamkeit beider lokalen Therapien direkt vergleichen. Die INT-0091-Studie zeigte keinen signifikanten Unterschied in Bezug auf lokale Rückfallraten oder ereignisfreies Überleben zwischen alleiniger Operation und alleiniger Strahlentherapie. Allerdings wurde eine Kombination aus Operation und RT mit einer niedrigeren Rate lokaler Rückfälle in Verbindung gebracht. Dennoch wird angenommen, dass ein Rezidiv bei alleiniger Strahlentherapie ohne Operation praktisch unvermeidlich ist. Daten von 1058 Patient:innen aus den Studien CESS 81, CESS 86 und EICESS-92 zeigten, dass Operation + RT sowie alleinige Operation signifikant niedrigere lokale Rückfallraten aufwiesen als alleinige Strahlentherapie. Die Inzidenz lokaler Rückfälle war in der präoperativen RT-Gruppe und in der chirurgischen Gruppe mit oder ohne postoperativer RT ähnlich. Daten aus den Studien INT-0091, INT-0154 und AEWS0031 belegen, dass Operation + RT mit einem geringeren Risiko für lokale Rückfälle assoziiert ist als alleinige Strahlentherapie.
2. Chondrosarkom
Behandlung des Chondrosarkoms: Symptome des Chondrosarkoms:
Was ist ein Chondrosarkom?
Ein Chondrosarkom ist eine Art von Knochenkrebs, der sich aus Knorpelzellen entwickelt. Knorpel ist ein spezialisiertes Bindegewebe, das bei Erwachsenen vorkommt und das Ausgangsgewebe für die Entwicklung vieler Knochen darstellt. Knorpel spielt eine wichtige Rolle im Wachstumsprozess. Es gibt viele verschiedene Arten von Knorpelgewebe im Körper. Chondrosarkome betreffen hauptsächlich die Knorpelzellen im Oberschenkelknochen (Femur), Oberarm, Becken oder Knie. Auch andere Bereiche (wie die Rippen) können betroffen sein, wenn auch seltener. Das Chondrosarkom ist die zweithäufigste Form des primären Knochentumors. Primärer Knochenkrebs ist Krebs, der direkt im Knochen entsteht. Diese Krebsart betrifft selten Menschen unter 20 Jahren. Das Risiko steigt kontinuierlich bis zum Alter von 75 Jahren. Die Inzidenz ist bei Männern und Frauen gleich.
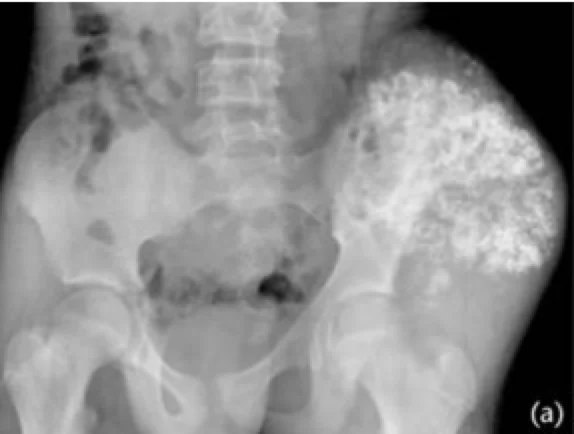
Was verursacht ein Chondrosarkom?
Die genaue Ursache des Chondrosarkoms ist nicht bekannt. Es kann eine genetische oder chromosomale Komponente geben, die manche Menschen anfälliger für solche bösartigen Erkrankungen macht. Chondrosarkome wurden auch als Spätfolge einer Strahlentherapie bei anderen Krebserkrankungen beobachtet.
Was sind die Risikofaktoren für ein Chondrosarkom?
In den meisten Fällen entsteht ein Chondrosarkom aus normalen Knorpelzellen. Es kann aber auch aus einem bereits bestehenden gutartigen (nicht-krebsartigen) Knochen- oder Knorpeltumor hervorgehen. Nachfolgend finden Sie eine Liste einiger gutartiger Erkrankungen, die mit dem Auftreten eines Chondrosarkoms in Verbindung stehen können:
- Enchondrome: Eine Art gutartiger Knochentumor, der im Knorpel entsteht und in der Regel die Hände betrifft (kann jedoch auch andere Bereiche betreffen).
- Multiple Exostosen (Osteochondrome): Vorhandensein mehrerer Osteochondrome.
- Ollier-Krankheit: Eine Ansammlung von Enchondromen (gutartige Knorpeltumoren, die meist die Hände betreffen).
- Maffucci-Syndrom: Eine Kombination aus multiplen Enchondromen (gutartige Knorpeltumoren, die meist die Hände betreffen) und Angiomen (gutartige Tumoren, die aus Blutgefäßen bestehen).
Was sind die Symptome eines Chondrosarkoms?
Die Symptome eines Chondrosarkoms können je nach Lage des Tumors variieren. Nachfolgend sind die häufigsten Symptome eines Chondrosarkoms aufgeführt. Allerdings können sich die Beschwerden bei jedem Individuum unterschiedlich äußern. Zu den möglichen Symptomen gehören:
- Große Raumforderung im betroffenen Knochen
- Druckgefühl rund um die Masse
- Schmerzen, die im Verlauf allmählich zunehmen. Sie verschlimmern sich meist nachts und können durch entzündungshemmende Medikamente wie Ibuprofen gelindert werden. Ruhe lindert die Beschwerden in der Regel nicht.
- Lokale Schwellung

Wie wird ein Chondrosarkom diagnostiziert?
Zusätzlich zur vollständigen Krankengeschichte und körperlichen Untersuchung können die folgenden diagnostischen Verfahren zur Anwendung kommen:
- Röntgenaufnahme: Eine diagnostische Methode, bei der unsichtbare elektromagnetische Strahlen genutzt werden, um Bilder des inneren Gewebes, der Knochen und Organe auf Film darzustellen.
- Computertomographie (auch CT oder CAT-Scan genannt): Ein bildgebendes Verfahren, bei dem Röntgenstrahlen und ein Computer verwendet werden, um detaillierte Querschnittsbilder des Körpers zu erstellen. Eine CT-Aufnahme zeigt Details von Knochen, Muskeln, Fett und Organen.
- Magnetresonanztomographie (MRT): Ein diagnostisches Verfahren, das eine Kombination aus starken Magnetfeldern, Radiowellen und Computern verwendet, um detaillierte Bilder von Organen und inneren Strukturen zu erstellen.
- Positronen-Emissions-Tomographie (PET): Ein bildgebendes Verfahren, bei dem radioaktiv markierte Glukose (Zucker) in den Blutkreislauf injiziert wird. Gewebe mit erhöhtem Glukoseverbrauch – wie Tumoren – können durch das Scangerät sichtbar gemacht werden.
- Biopsie: Ein Verfahren, bei dem Gewebeproben (mittels Nadel oder während einer Operation) aus dem Körper entnommen und unter dem Mikroskop untersucht werden. Dies dient dem Nachweis von Krebszellen oder anderen krankhaften Zellveränderungen.
Behandlung des Chondrosarkoms:
Die spezifische Behandlung eines Chondrosarkoms wird von Ihrem behandelnden Arzt unter Berücksichtigung folgender Faktoren festgelegt:
- Ihr Alter, allgemeiner Gesundheitszustand und Ihre Krankengeschichte
- Art, Stadium (Ausbreitung) und Lage des Tumors
- Ihre Verträglichkeit gegenüber bestimmten Medikamenten, Verfahren und Therapien
- Die zu erwartende Entwicklung der Erkrankung
- Ihre persönliche Meinung oder Präferenz
Ziel der Behandlung ist es, die Raumforderung vollständig zu entfernen und die Wahrscheinlichkeit eines Rückfalls zu minimieren. Die Behandlung kann folgende Maßnahmen umfassen:
- Operation: Entfernung des Tumors. Wenn sich der Tumor in einem Arm oder Bein befindet, wird der Chirurg versuchen, die Extremität zu erhalten. In einigen Fällen kann jedoch eine Amputation erforderlich sein.
- Physiotherapie: Diese Maßnahme hilft dabei, nach der Operation die Kraft und Funktion des betroffenen Bereichs wiederzuerlangen.
- Strahlentherapie: Die Bestrahlung kann in hohen Dosen verabreicht werden.
- Chemotherapie: Obwohl sie keine primäre Behandlung darstellt, kann sie erforderlich sein, wenn sich der Tumor auf andere Körperteile ausgebreitet hat.
3. Osteosarkom
Was ist ein Osteosarkom?
Ein Osteosarkom (OS) oder osteogenes Sarkom (OGS) – auch als Knochenkrebs bezeichnet – ist ein bösartiger Tumor im Knochen. Genauer gesagt handelt es sich um eine aggressive maligne Neoplasie, die aus primitiven, transformierten Zellen mesenchymalen Ursprungs hervorgeht, osteoblastische Differenzierung zeigt und malignen Osteoid produziert. Das Osteosarkom ist die häufigste histologische Form des primären Knochensarkoms. Es tritt am häufigsten bei Jugendlichen und jungen Erwachsenen auf.
Symptome des Osteosarkoms:
Viele Patient:innen klagen zunächst über anhaltende Schmerzen, die sich nachts verschlimmern, intermittierend auftreten und unterschiedlich stark ausgeprägt sein können. Besonders junge Menschen, die aktiv Sport treiben, berichten häufig über Schmerzen im unteren Teil des Oberschenkelknochens oder knapp unterhalb des Knies. Wenn der Tumor groß ist, kann er sich als deutliche lokale Schwellung äußern. Mitunter ist eine plötzliche Fraktur das erste Symptom, da der betroffene Knochen nicht mehr so stabil ist wie ein gesunder Knochen und schon bei geringer Belastung brechen kann. Bei Tumoren, die tiefer im Körper liegen und nicht direkt unter der Haut sind – wie etwa im Beckenbereich – kann eine lokale Schwellung unter Umständen nicht sichtbar sein.
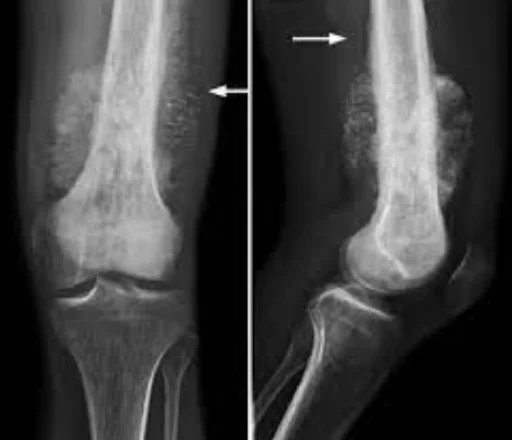
Ursachen des Osteosarkoms:
Familiäre Fälle, bei denen eine Deletion des Chromosoms 13q14 das Retinoblastom-Gen inaktiviert, sind mit einem hohen Risiko für die Entwicklung eines Osteosarkoms verbunden. Knochendysplasien wie die Paget-Krankheit, fibröse Dysplasie, Enchondromatose und hereditäre multiple Exostosen erhöhen das Osteosarkomrisiko.
Das Li-Fraumeni-Syndrom (Keimbahnmutation des TP53-Gens) ist ein prädisponierender Faktor für die Entwicklung eines Osteosarkoms. Das Rothmund-Thomson-Syndrom (eine autosomal-rezessiv vererbte Kombination aus angeborenen Knochendefekten, Haar- und Hautdysplasien, Hypogonadismus und Katarakt) ist ebenfalls mit einem erhöhten Risiko für diese Erkrankung verbunden. Hohe Dosen von Strontium-90 (Sr-90) erhöhen das Risiko für Knochentumoren und Leukämien bei Tieren und gelten auch beim Menschen als risikobehaftet.
Verursacht Fluorid ein Osteosarkom?
Es besteht kein klarer Zusammenhang zwischen der Fluoridierung von Trinkwasser und Krebs oder krebsbedingten Todesfällen – weder allgemein noch spezifisch in Bezug auf Knochentumoren oder Osteosarkome. Eine Reihe von Studien kam zu dem Schluss, dass die Fluoridkonzentration im Trinkwasser nicht mit Osteosarkomen assoziiert ist. Der Glaube an einen Zusammenhang zwischen Fluoridexposition und Osteosarkom geht auf eine Studie des US-amerikanischen National Toxicology Program aus dem Jahr 1990 zurück, die bei männlichen Ratten einen uneindeutigen Hinweis auf einen Zusammenhang zeigte. Es gibt jedoch bis heute keine stichhaltigen Beweise dafür, dass Fluorid Krebs bei Mäusen verursacht. Die Fluoridierung von Trinkwasser wird weltweit zur Förderung der Zahngesundheit praktiziert und gilt als bedeutender medizinischer Fortschritt. Der Fluoridgehalt im Wasser wird reguliert – beispielsweise darf er laut der US-Umweltschutzbehörde (EPA) 4 mg/L nicht überschreiten. Tatsächlich ist Fluorid natürlicherweise bereits im Wasser enthalten; viele Gemeinden fügen jedoch zusätzlich Fluorid hinzu, um Karies vorzubeugen. Fluorid ist auch dafür bekannt, die Knochenneubildung zu fördern. Weitere Untersuchungen zeigen jedoch, dass fluoridiertes Wasser kein erhöhtes Osteosarkomrisiko beim Menschen darstellt. Die meisten Studien analysierten die Anzahl der Osteosarkomfälle in Regionen mit unterschiedlichen Fluoridkonzentrationen im Trinkwasser. Statistische Auswertungen ergaben keine signifikanten Unterschiede in der Inzidenzrate. Eine weitere wichtige Studie verglich die Fluoridkonzentration in Knochenproben von Osteosarkompatienten mit Proben anderer maligner Knochentumoren. Das Ergebnis: Die durchschnittlichen Fluoridkonzentrationen unterschieden sich nicht signifikant. Es wurde nachgewiesen, dass weder die Fluoridmenge in den Knochen noch die Exposition gegenüber Fluorid bei Osteosarkompatienten im Vergleich zu gesunden Personen signifikant unterschiedlich ist.
Osteosarkome treten bevorzugt in Wachstumszonen der Knochen auf – wahrscheinlich, weil sich in diesen Regionen osteoblastische Zellen verstärkt teilen und somit anfälliger für Mutationen sind (z. B. im RB- oder p53-Gen). Der Tumor befindet sich meist am Ende langer Röhrenknochen, typischerweise in der Metaphyse. Am häufigsten betroffen sind der proximale Teil der Tibia oder des Humerus sowie das distale Femurende. In etwa 60 % der Fälle ist das Kniegelenk betroffen, in 15 % die Hüfte, in 10 % die Schulter und in 8 % der Kiefer. Der Tumor erscheint als feste, harte und unregelmäßige Raumforderung, da die Tumorstrahlen im verkalkten Knochen rechtwinklig abstrahlen (sog. „Tannenbaum-“, „Mottenfraß-“ oder „Sonnenuntergangs“-Muster in der Röntgenaufnahme). Diese rechtwinkligen Ausläufer formen das sogenannte Codman-Dreieck – ein charakteristisches, jedoch nicht beweisendes Merkmal eines Osteosarkoms.
Diagnose des Osteosarkoms:
Röntgenaufnahmen sind die bevorzugte bildgebende Erstmaßnahme zur Diagnose eines Osteosarkoms. Typische Merkmale auf dem Röntgenbild sind das sogenannte „Sonnenuntergangs-Phänomen“ sowie das Codman-Dreieck (eine Anhebung der Knochenkortikalis durch den Tumor, die zur Neubildung von Knochen führt). Die CT (Computertomographie) ist hilfreich zur Darstellung der Knochenanatomie, zur Beurteilung der Integrität der Kortikalis, zum Nachweis pathologischer Frakturen sowie zur Einschätzung von Ossifikationen (Neubildung von Knochengewebe) und Knorpelverkalkungen. Die MRT (Magnetresonanztomographie) hingegen bietet eine bessere Darstellung der Weichteile und des Markraums.
Eine Biopsie bei Verdacht auf ein Osteosarkom sollte unbedingt von einem qualifizierten orthopädischen Onkologen durchgeführt werden. Die American Cancer Society betont: „Wahrscheinlich ist es bei keiner anderen Krebsart wichtiger, dass dieser Eingriff korrekt durchgeführt wird.“ Eine unsachgemäße Biopsie kann die Chance auf Gliedmaßenerhalt deutlich verringern und im schlimmsten Fall zur Amputation führen. Außerdem kann das Osteosarkom in die Lunge metastasieren – häufig sichtbar als einzelne oder multiple runde Knoten in den unteren Lungenfeldern auf dem Thoraxröntgenbild.
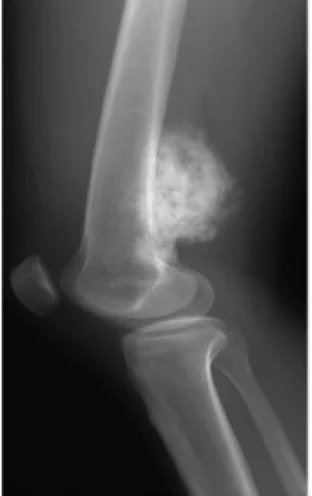
Osteosarkom-Typen:
- Konventionell: osteoblastisch, chondroblastisch, fibroblastisch
- Telangiektatisch
- Kleinzellig
- Niedriggradig zentral
- Periostal
- Parostal
- Sekundär
- Hochgradig oberflächlich
- Extraskelettal
Behandlung des Osteosarkoms:
Die vollständige chirurgische Resektion des Tumors im Sinne einer en-bloc-Entfernung ist die bevorzugte Behandlungsmethode bei Osteosarkomen. Obwohl bei den meisten Patient:innen ein gliedmaßenerhaltender Eingriff möglich ist, können Komplikationen – insbesondere Infektionen, Lockerung der Prothese, Pseudarthrose oder ein lokales Tumorrezidiv – weitere Operationen oder eine Amputation erforderlich machen.
PATIENT:INNEN MIT OSTEOSARKOM SOLLTEN VON EINEM ORTHOPÄDISCHEN ONKOLOGEN BEHANDELT WERDEN, DER AUF DIE THERAPIE VON SARKOMEN SPEZIALISIERT IST. Der derzeitige Behandlungsstandard ist eine neoadjuvante Chemotherapie (vor der Operation verabreichte Chemotherapie), gefolgt von einer chirurgischen Resektion. Der Prozentsatz der Tumornekrose (Zelluntergang), der im Tumor nach der Operation festgestellt wird, gibt Hinweise auf die Prognose und hilft dem Onkologen dabei zu entscheiden, ob das Chemotherapieprotokoll angepasst werden muss.
Der Standardansatz ist möglichst eine gliedmaßenerhaltende orthopädische Operation (oder in einigen Fällen eine Amputation), in Kombination mit einer Hochdosis-Methotrexattherapie und Leucovorin-Rescue sowie einer Kombination aus intraarteriellem Cisplatin, Adriamycin, Mesna mit Ifosfamid, BCD (Bleomycin, Cyclophosphamid, Dactinomycin), Etoposid und Muramyl-Tripeptid. Das verwendete Protokoll ist ein aggressives intraarterielles Regime, das die Behandlung anhand der arteriographischen Reaktion individualisiert. In einigen Studien liegt das ereignisfreie 3-Jahres-Überleben bei 50 % bis 75 %, das 5-Jahres-Überleben bei 60 % bis 85 %. Insgesamt sind 65 % bis 70 % der Patient:innen, die vor fünf Jahren behandelt wurden, heute noch am Leben. Diese Überlebensraten stellen Durchschnittswerte dar und hängen stark vom individuellen Nekrosegrad ab.
Krankheitsverlauf des Osteosarkoms:
Ein Osteosarkom Grad I ist selten und umfasst das parostale Osteosarkom und das niedriggradig zentrale Osteosarkom. Diese Formen haben bei vollständiger Resektion eine exzellente Prognose (>90 %). Die Gesamtprognose hängt vom Ort des Tumors (z. B. proximale Tibia, Femur, Becken), der Größe der Raumforderung sowie vom Grad der Tumornekrose nach neoadjuvanter Chemotherapie ab. Weitere pathologische Faktoren, wie der P-Glykoprotein-Ausdruck, CXCR4- oder HER2-Positivität, sind ebenfalls bedeutsam, da sie mit dem Auftreten von Fernmetastasen – insbesondere in der Lunge – assoziiert sind. Die Mortalitätsraten beim Osteosarkom sinken jährlich um etwa 1,3 %. Die Langzeitüberlebenschancen haben sich insbesondere im späten 20. Jahrhundert deutlich verbessert und lagen im Jahr 2009 bei etwa 68 %.
