




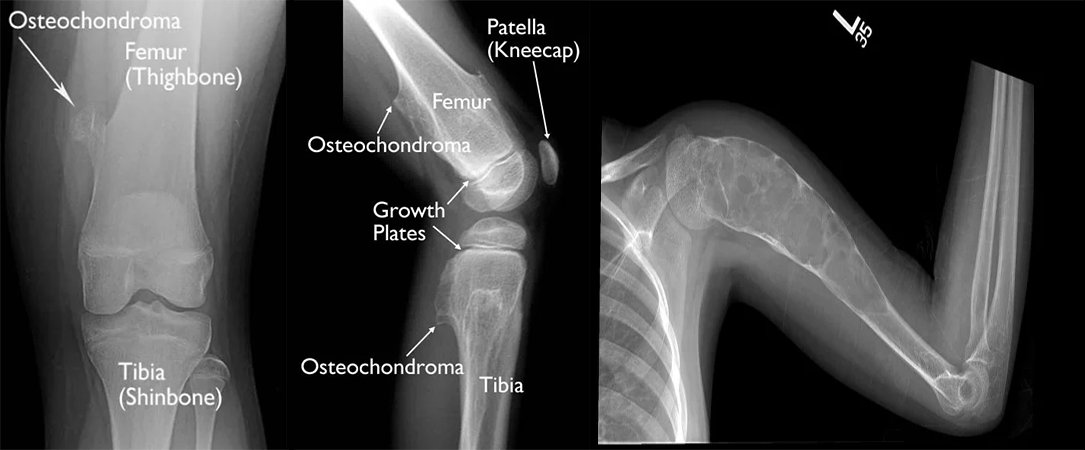
Osteochondrom
Was ist ein Osteochondrom? – Osteochondrom-Operation
Ein Osteochondrom ist ein gutartiger, nicht krebsartiger Knochentumor, der sich im Kindes- und Jugendalter entwickelt. Es handelt sich um eine abnorme Entwicklung an der Knochenoberfläche im Bereich der Wachstumsfuge. Wachstumsfugen ermöglichen die Knorpelbildung an den Enden langer Knochen. Das Knochenwachstum erfolgt durch die Wachstumsfuge. Nach der vollständigen Entwicklung des Kindes verhärten sich die Wachstumsfugen und entwickeln sich zu normalem Knochen. Ein Osteochondrom ist ein Gewebe aus Knochen und Knorpel, das durch die übermäßige Entwicklung der Wachstumsfuge entsteht. Es ist normal, dass Osteochondrome während der Skelettentwicklung des Kindes wachsen. Nach Abschluss der Skelettentwicklung stoppt das Osteochondrom jedoch sein Wachstum.
Behandlung von Osteochondromen
In vielen Fällen ist bei Osteochondromen außer regelmäßigen Kontrolluntersuchungen keine Behandlung erforderlich. Sollten sich jedoch Veränderungen im Gewebe der Raumforderung oder Komplikationen aufgrund einer Gefäßnervenkompression zeigen, kann eine Operation erforderlich sein. Osteochondrome können sich als einzelner Tumor (osteokartilaginäre Exostose) oder als multiple Tumore (multiple Osteochondromatose) entwickeln.
Chirurgische Behandlung von Osteochondromen
Ihr Arzt wird eine Operation in Erwägung ziehen, wenn Sie folgende Symptome haben:
- Schmerzen
- Nerven- oder Gefäßkompression
- Vorhandensein einer breiten Knorpelkappe
Während der Operation wird die Masse bis auf das normale Knochenniveau entfernt.
Erholungszeit nach einer Osteochondrom-Operation: Die Rückkehr zu alltäglichen Aktivitäten dauert in der Regel 3–6 Wochen, variiert jedoch je nach Größe und Lage der Masse.
Solitäres Osteochondrom: Es ist der häufigste Knochentumor und macht 40 % aller Knochentumoren aus. Es handelt sich nicht um Krebs und streut nicht weit.
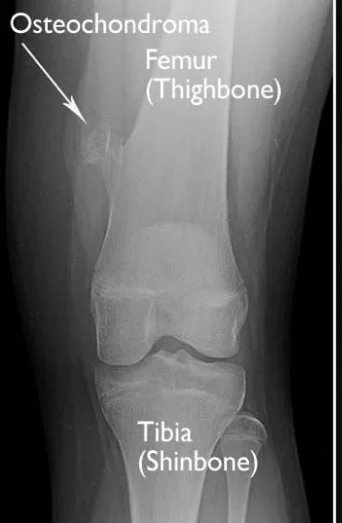
Die Ursache ist unbekannt. Es liegt nicht an einer Verletzung, sondern wird mit dem Gen EXT 1 in Zusammenhang gebracht. Da die Ursache unbekannt ist, ist eine Vorbeugung nicht möglich. Osteochondrome werden häufig im Alter zwischen 10 und 30 Jahren diagnostiziert.
Multiple Osteochondromatose: Andere Bezeichnungen sind multiple Exostose, multiple hereditäre Exostose, familiäre Osteochondromatose, multiple hereditäre Osteochondromatose und diaphysäre Achalasie. In fortgeschrittenen Fällen können Knochenentwicklungsstörungen, Kleinwuchs und Unterarmdeformitäten auftreten.
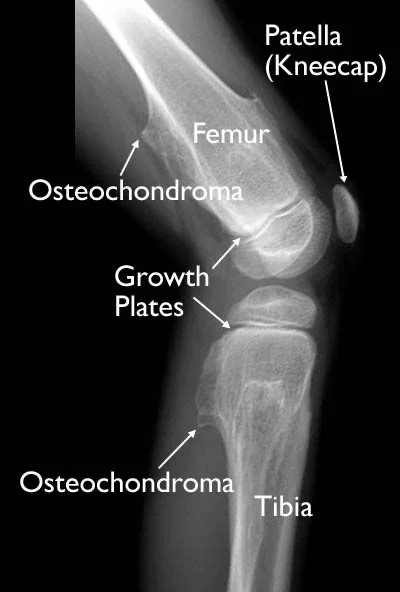
Bei diesem Syndrom ist die Entwicklung einer gutartigen Raumforderung zu einer Verschlechterung häufiger als die Entwicklung einzelner Osteochondrome. Während die Ursache in 70 % der Fälle familiär-genetisch bedingt ist, entwickelt sie sich in 30 % der Fälle spontan.
Orthopädische onkologische Untersuchung: Wenn bei Ihnen oder Ihrem Kind Anzeichen einer Krebserkrankung auftreten, sollten Sie einen auf Knochentumoren spezialisierten Orthopäden aufsuchen.
Krebssymptome:
- Wachstum eines Osteochondroms nach der Pubertät
- Schmerzen im Bereich der Masse
- Eine Knorpelkappe breiter als 2 cm
In diesem Fall sollten MRT und CT durchgeführt werden. Gegebenenfalls wird bei Verdacht auf Metastasierung eine Lungen-CT durchgeführt. Falls Ihr Arzt es für notwendig erachtet, kann eine Biopsie der Masse die Krebsdiagnose unterstützen. Entsteht Krebs, handelt es sich häufig um ein Chondrosarkom.
Aneurysmatische Knochenzyste – Einfache Knochenzyste
Was ist eine Knochenzyste? Behandlung von Knochenzysten: Einfache Knochenzyste: Es handelt sich um eine häufige gutartige Knochenläsion. Sie tritt häufig bei Kindern auf und verläuft in der Regel asymptomatisch. Sie treten in der Regel erstmals im Alter von 20 Jahren auf. 65 % der Fälle betreffen Kinder unter 18 Jahren, das durchschnittliche Diagnosealter liegt bei 9 Jahren. Jungen sind zwei- bis dreimal häufiger betroffen als Mädchen. Aktive Zysten treten häufig im Alter zwischen 1 und 10 Jahren auf. Diese Zysten verursachen oft keine Symptome und werden zufällig entdeckt. In einigen Fällen können jedoch Schmerzen, Schwellungen und Bewegungseinschränkungen im benachbarten Gelenk auftreten. Die häufigste Überweisung zum Arzt erfolgt aufgrund eines unerwarteten Bruchs. Ursache und Mechanismus der Erkrankung sind unbekannt. In der aktiven Phase der Erkrankung grenzt die Zyste an die Wachstumsfuge und entfernt sich mit zunehmender Inaktivität von der Wachstumsfuge. Das bedeutet, dass sich zwischen Wachstumsfuge und Zyste normaler Knochen bildet. Die Zyste verschwindet in der Regel mit zunehmendem Alter. Während die Diagnose einer einfachen Knochenzyste üblicherweise durch direkte Röntgenaufnahmen gestellt wird, können Tomographie und MRT zur Differentialdiagnose erforderlich sein. Einfache Knochenzysten liegen typischerweise intramedullär. Aktive Zysten befinden sich im metaphysären Bereich der langen Knochen und ruhen auf der Wachstumsfuge.
- Der proximale Humerus ist mit 50–60 % am häufigsten betroffen.
- Der proximale Femur mit 30 %.
- Andere lange Röhrenknochen.
Andere Lokalisationen sind selten und treten meist bei Erwachsenen auf.
- Die Wirbelsäule bildet meist die hinteren Elemente.
- Das Becken hat nur einen Anteil von 2 %.
- Calcaneus-Fersenbein.
Im Röntgenbild sind die Läsionen transparent und geographisch gut abgegrenzt. Es ist eine schmale Übergangszone erkennbar. Sie tritt meist bei Patienten mit unvollständiger Skelettentwicklung auf. Sie befindet sich im Zentrum des Knochens. Es besteht ein dünner sklerotischer Rand. Es sind keine Periostreaktionen oder Weichteilanteile erkennbar. Sie dehnen den Knochen in der Regel aus und verdünnen die Knochenmembran. Liegt jedoch keine pathologische Fraktur vor, perforieren sie die Kortikalis nicht. Einfache Knochenzysten sind oft unilokulär, obwohl Pseudotrabekel auftreten können. Echte Trabekel sind nur bei wiederkehrenden Frakturen sichtbar. Liegt innerhalb der Läsion eine Fraktur vor, ist ein loses Knochenfragment sichtbar; dieses Bild wird als „fallendes Blattzeichen“ bezeichnet.
CT: Sie eignet sich zur Erkennung von Frakturen, die im Graphit nicht direkt sichtbar sind, und kann auch zur Beurteilung des Zysteninhalts eingesetzt werden.
Emar: Flüssigkeitsspiegel können sichtbar gemacht werden. Bei einer Fraktur können Signalheterogenität, Periostreaktionen und Weichteilödeme auftreten.
Behandlung einfacher Knochenzysten: Bei asymptomatischen Läsionen ist in der Regel keine Behandlung erforderlich. Bei sehr großen Läsionen mit Frakturrisiko oder Deformität kann eine intrazystische Steroidinjektion in die Arme erfolgen. Befindet sich die Zyste im Hüftknochen, können Kürettage und Knochentransplantation erforderlich sein. Die Zyste unterscheidet sich von NOF und aneurysmatischen Knochenzysten durch ihre zentrale Lage und von Riesenzelltumoren dadurch, dass die Zyste die Gelenkoberfläche nicht erreicht und in jungen Jahren sichtbar wird. Darüber hinaus kann es notwendig sein, eine Differentialdiagnose mit fibrösen Dysplasiezysten, intraossärem Lipom und Ganglion zu stellen.
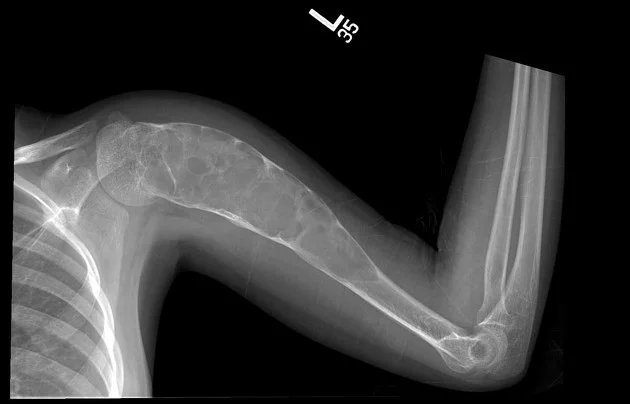
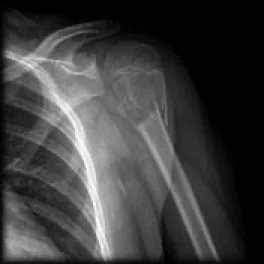
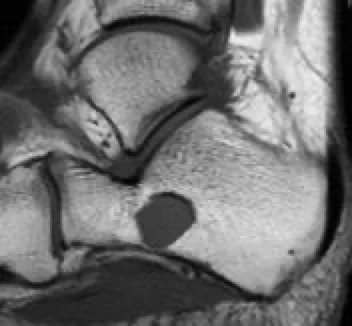
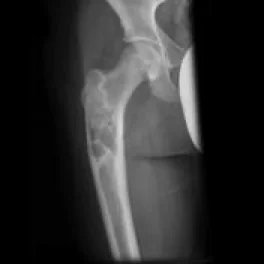
Aneuryzmatische Knochenschichtzyste: Operation einer Knochenzyste:
Es handelt sich um gutartige, expansive, lythische, zystische Raumforderungen, die reich an osteoklastären Riesenzellen sind. Sie treten häufig bei Kindern und Jugendlichen auf. 80 % der Fälle betreffen Personen unter 20 Jahren, können aber in jedem Alter vorkommen. Es gibt keinen Geschlechtsunterschied. Die Diagnose erfolgt durch Bildgebung und Biopsie.
- Multizystische Raumforderung mit Flüssigkeitsspiegeln
- Pathologie: Zystenwände enthalten fibroblastäre Zellen, osteoklastäre Riesenzellen, Hämosiderinpigment und neue Knochenbildung
- In 60 % der Fälle ist das USP6-Gen nachweisbar
Symptome einer aneurysmatischen Knochenzyste: Schmerzen und Schwellung. In seltenen Fällen kann eine pathologische Fraktur auftreten. Wenn sie sich in der Wirbelsäule befindet, kann sie eine Nervenkompression verursachen. Die Lokalisation liegt meist in der Metaphyse des Knochens, exzentrisch, angrenzend an die Wachstumsfuge.
- Am häufigsten in langen Röhrenknochen (65 %), insbesondere Femur, proximale Tibia und Fibula sowie Humerus
- In der Wirbelsäule und im Becken (20–30 %). In 40 % der Fälle ragen die hinteren Elemente in den Wirbelkörper oder in die Obturatorregion des Beckens
Bei Lokalisation in kurzen Knochen befindet sich die Zyste meist zentral.
Obwohl Flüssigkeitsspiegel im MRT charakteristisch für aneurysmatische Knochenzysten sind, sind sie niemals pathognomonisch. Es ist wichtig zu beachten, dass solche Flüssigkeitsspiegel sowohl bei gutartigen (z. B. Riesenzelltumor, Chondroblastom, einfache Knochenzyste) als auch bei bösartigen Tumoren (z. B. telangiektatisches Osteosarkom) vorkommen können.
Behandlung der aneurysmatischen Knochenzyste: Therapie gutartiger Knochentumoren:
Obwohl es sich um gutartige Tumoren handelt, können sie unterschiedliche klinische Verläufe zeigen: Sie können stumm (asymptomatisch), aktiv oder aggressiv sein. Die Behandlung erfolgt in der Regel durch Kürettage (Ausschabung) oder Exzision (vollständige Entfernung) und anschließendem Knochengrafting (Einbringung von Knochenspänen). Bei geschwächten Knochen wird zusätzlich eine Stabilisierung mit Metallplatten, Nägeln oder Schrauben durchgeführt.
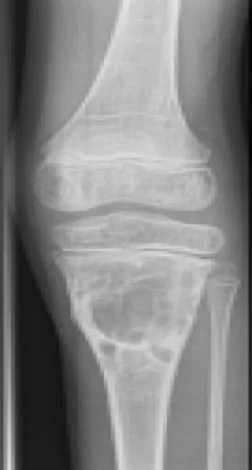
Abbildung 7: Aneurysmatische Knochenzyste am oberen Ende der Tibia.
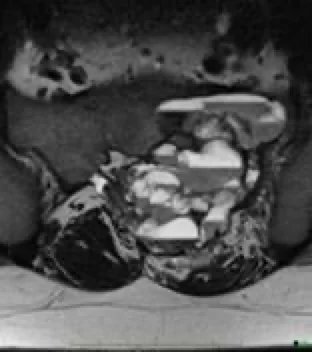
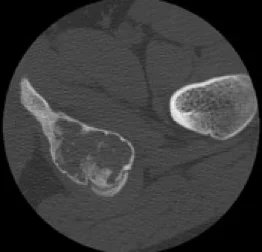
Abbildung 9: Aneurysmatische Knochenzyste im Becken.
Das Rezidivrisiko kann je nach Behandlungsmethode auf bis zu 30 % steigen.
Osteoidosteom
Symptome eines Beintumors: Behandlung des Osteoidosteoms: Osteoidosteome sind gutartige, knochenbildende Tumoren, die meist bei Kindern (insbesondere Jugendlichen) auftreten. Sie besitzen einen charakteristischen Nidus (1,5 bis 2 cm) mit umgebender osteosklerotischer Reaktion und verursachen typischerweise nächtliche Schmerzen, die durch NSAR (Schmerzmittel) gelindert werden. Das Osteoidosteom tritt überwiegend bei Kindern, Jugendlichen und jungen Erwachsenen im Alter von 10 bis 35 Jahren auf. Es macht etwa 10 % aller gutartigen Knochenläsionen aus und ist bei Männern 2- bis 4-mal häufiger als bei Frauen. Es ist die klassische Ursache für eine schmerzhafte Skoliose der Wirbelsäule, bei der die Krümmung auf der Seite der Läsion konkav ist – dieses typische Erscheinungsbild wird in über 75 % der Wirbelfälle beobachtet. Eine Weichteilschwellung durch den Tumor kann auftreten. Befindet sich der Tumor in der Nähe der Wachstumsfuge, kann auf dieser Seite durch Hyperämie ein beschleunigtes Wachstum erfolgen. Liegt die Läsion innerhalb der Gelenkkapsel, kann sie eine Gelenkentzündung (inflammatorische Arthropathie oder Synovitis) imitieren. In solchen Fällen liegt häufig ein Gelenkerguss vor.
In welchem Knochen tritt ein Osteoidosteom auf? Die meisten Osteoidosteome treten in den langen Röhrenknochen der Extremitäten auf (insbesondere im proximalen Femur), aber grundsätzlich kann jeder Knochen betroffen sein.
- Lange Röhrenknochen der Gliedmaßen: ~65–80 %
- Am häufigsten Femur (insbesondere Schenkelhals)
- Mittlere Tibia-Diaphyse
- Phalangen: ~20 %
- Wirbelkörper: ~10 %, überwiegend hintere Elemente
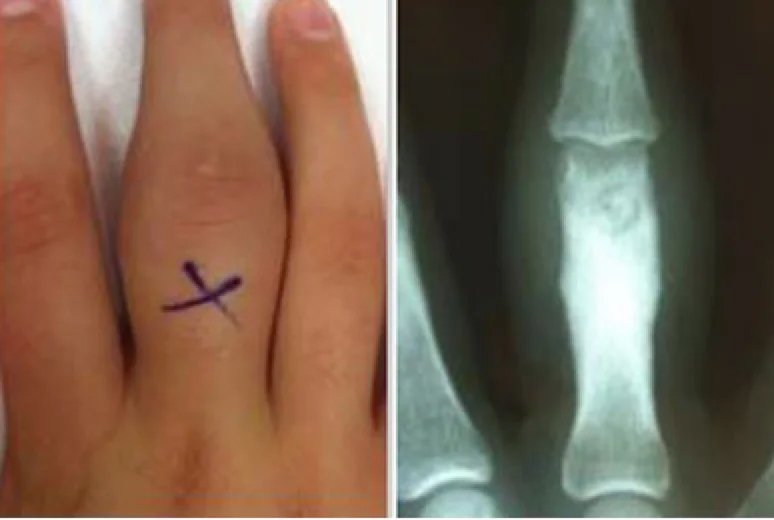
Osteoidosteome sind in der Regel kortikale Läsionen, können jedoch überall im Knochen auftreten – einschließlich im Markraum (medullär), subperiostal (am häufigsten im Talus) oder intrakapsulär. Bei intrakapsulären Osteoidosteomen kann die periostale Reaktion entfernt vom eigentlichen Herd auftreten.
Diagnose eines Osteoidosteoms: Konventionelle Röntgenaufnahme:
Die Röntgenaufnahme kann unauffällig sein oder eine solide periostale Reaktion mit kortikaler Verdickung zeigen. Der Nidus kann manchmal als klar umgrenzte Aufhellung sichtbar sein, gelegentlich mit einem zentralen sklerotischen Punkt. Eine ausgeprägte Sklerosierung kann diesen Fokus jedoch gelegentlich überdecken.
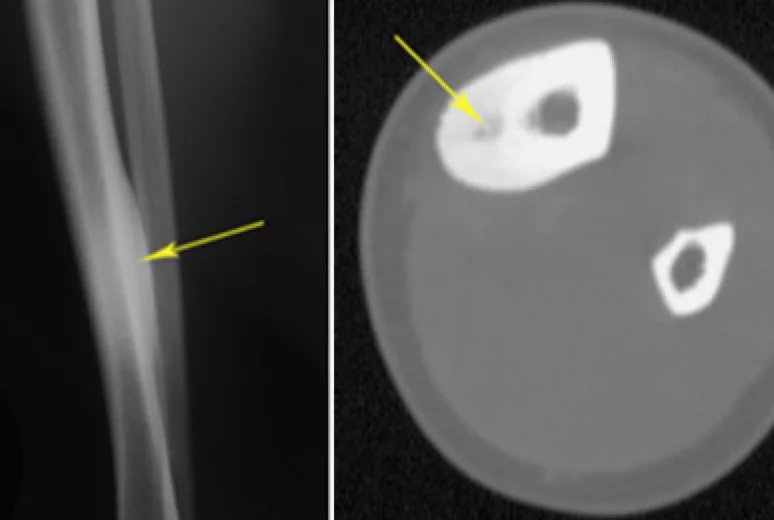
BT:
Die CT eignet sich hervorragend zur Charakterisierung der Läsion und ist die Methode der Wahl. Sie zeigt typischerweise einen fokalen, transparenten Fleck im umgebenden sklerotischen, reaktiven Knochen. Ein zentraler sklerotischer Fleck kann ebenfalls sichtbar sein.
MRT:
Die MRT ist zwar sensitiv, aber unspezifisch und kann den Fokus oft nicht identifizieren. Das Muster der Hyperämie und das daraus resultierende Knochenmarködem können dazu führen, dass Scans als aggressive Pathologie fehlinterpretiert werden. Die Signalintensität des Nidus und der Grad der Kontrastverstärkung sind in allen Sequenzen variabel. Bei Patienten ohne Überbeanspruchung der unteren Extremitäten in der Anamnese ist die Bestimmung des Halbmondzeichens hochspezifisch und sensitiv für die Erkennung eines Osteoidosteoms des Schenkelhalses. In diesen Fällen muss das Halbmondzeichen in flüssigkeitssensitiven MRT-Sequenzen nachgewiesen werden. Bei Überbeanspruchung (z. B. Marathonläufer, Angehörige der Streitkräfte) kann das Halbmondzeichen auf eine Belastungsreaktion/Fraktur hinweisen.
Nuklearmedizin:
Bei der Knochenszintigraphie zeigt sich eine typische fokale Kontrastverstärkung und gelegentlich ein Doppeldichtezeichen (auch als weniger heißer Punkt innerhalb eines heißen Bereichs bezeichnet). Dieses ist, wenn vorhanden, hochspezifisch und hilft bei der Unterscheidung zwischen Osteoidosteom und Osteomyelitis.
Behandlung von Osteoidosteomen:
Die Läsion ist gutartig und wird traditionell durch chirurgische Resektion behandelt. In der Vergangenheit war dies manchmal schwierig, da der Nidus zum Zeitpunkt der Operation nicht lokalisiert werden konnte. In ausgewählten Fällen werden anstelle einer Operation auch perkutane Methoden eingesetzt.
Mit Osteoidosteom verwechselte Läsionen: Differentialdiagnose
- Osteomyelitis (z. B. Brodie-Abszess): Eine Knochenszintigraphie zeigt einen zentralen Bereich verminderter Aufnahme, der einen avaskulären Bereich eitrigen Materials darstellt.
- Osteoblastom: >1,5–2 cm groß
- Stressfraktur
- Kortikales Desmoid
- Osteochondrom
- Osteosarkom
- Knocheninsel
- Lokalisierte kortikale Verdickung
- Intrakortikales Hämangiom
- Reaktive Sklerose um die osteolytische Läsion
Enchondrom
Enchondrome, auch Chondrome genannt, sind häufige intramedulläre hyaline Knorpelneoplasien, die in der Bildgebung gutartige Tumoreigenschaften aufweisen. Sie ähneln histologischen Merkmalen dem niedriggradigen Chondrosarkom und werden manchmal unter dem Oberbegriff niedriggradige Chondrale Tumoren zusammengefasst. Die Diagnose erfolgt am häufigsten im Kindes- und frühen Erwachsenenalter, die höchste Inzidenz tritt zwischen dem 10. und 30. Lebensjahr auf. Es handelt sich um den häufigsten primären gutartigen Knochentumor der Hand und des Handgelenks. Er macht etwa 5 % aller Knochentumoren und etwa 20 % der gutartigen Knochentumoren aus.
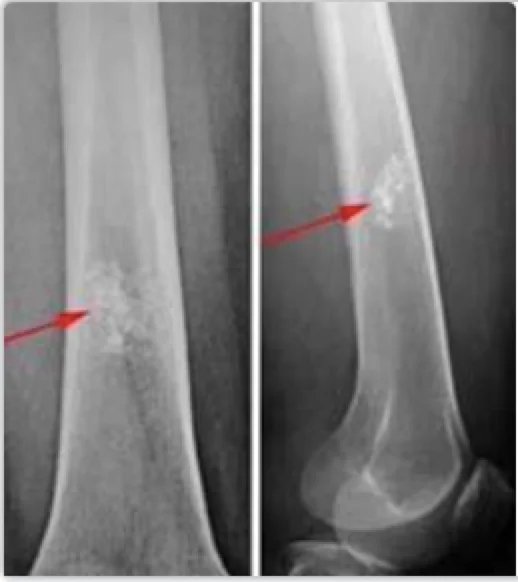
Diagnose eines Enchondroms:
Das Vorhandensein einer reichlich vorhandenen Knorpelmatrix und das Fehlen von Zellatypien/Mitosen, Weichteilausdehnung und kortikaler Invasion sind diagnostische Kriterien.
Symptome eines Enchondroms:
Enchondrome werden oft zufällig entdeckt. Dies ist besonders wichtig, um den Tumor nicht mit aggressiveren Läsionen zu verwechseln. In der Regel verlaufen Enchondrome asymptomatisch. Läsionen an Händen und Füßen können jedoch mit pathologischen Frakturen oder Schmerzen aufgrund einer drohenden Fraktur einhergehen. Eine maligne Transformation zu einem niedriggradigen Chondrosarkom ist selten und kann, wenn sie auftritt, mit Schmerzen einhergehen.
Enchondrom-Pathologie:
Enchondrome bestehen aus Läppchen reifen hyalinen Knorpels, die teilweise oder vollständig von normalem Knochen umgeben sind. Knorpelläppchen können enchondrale Ossifikation erfahren, was häufig zu dem charakteristischen Mineralisierungsmuster „Ringe und Bögen“ führt. Sie entstehen aus Wachstumsfugenknorpeln/Chondrozyten, die sich im reifen Knochen isoliert haben. Daher sind sie in jedem Knorpelknochen nachweisbar. Definitionsgemäß zeigen sie histologisch keine lokalen Hinweise auf eine Invasion (Differenzierung zum niedriggradigen Chondrosarkom). Es ist jedoch wichtig zu wissen, dass sich Enchondrome histologisch nicht zuverlässig von Chondrosarkomen unterscheiden lassen und die Diagnose auf der Korrelation klinischer, bildgebender und pathologischer Befunde beruht. Die Läsionen sind in der Regel 3 cm groß, durchscheinend, knotig und überwiegend graublau. Enchondrome treten typischerweise zentral oder exzentrisch im Markraum von Röhrenknochen auf:
- Kleine Röhrenknochen von Händen und Füßen (ca. 50 %)
- Am häufigsten ist die proximale Phalanx betroffen
- Große Röhrenknochen
Zum Beispiel Femur, Tibia, Humerus
In seltenen Fällen kann sich ein Enchondrom über die gesamte Kortikalis ausdehnen und ein exophytisches Wachstumsmuster aufweisen. Dies wird als Enchondroma protuberans bezeichnet und kann sporadisch oder im Rahmen der Ollier-Krankheit auftreten.

Röntgenbild und CT:
Enchondrome haben ein variables Erscheinungsbild, sind aber typischerweise kleine, 5 cm große intramedulläre lytische Läsionen mit nicht aggressiven Merkmalen:
- Schmale Übergangszone
- Scharf abgegrenzte Ränder
- +/- chondroide Verkalkung (Ring- und Bogenverkalkung)
In Händen und Füßen fehlt häufig die Matrixverkalkung (rein lythisch).
- +/- Expansion
- Häufiger in Händen und Füßen, seltener in langen Röhrenknochen (Tibia, Femur)
- Es kann zu leichter endostaler Aufrauung kommen
- Die Läsion sollte nicht durch die Kortikalis „wachsen“ (außer bei pathologischer Fraktur)
- Keine ausgeprägte Knochenzerstörung
- Keine periostale Reaktion
- Keine Weichteilmasse
Die meisten Enchondrome treten häufiger in der metaphysären Region auf, vermutlich weil sie von der Wachstumsfuge ausgehen. In der Epiphyse sind sie selten; ein Knorpeltumor in der Epiphyse ist mit größerer Wahrscheinlichkeit ein Chondrosarkom. Die MRT ist hilfreich zur Beurteilung der Weichteilinvasion und zur Bestätigung der Diagnose. Enchondrome erscheinen als gut umschriebene, leicht gelappte Massen, die das Knochenmark verdrängen. Die Abgrenzung zwischen Enchondrom und niedriggradigem Chondrosarkom ist problematisch, da beide ein ähnliches Erscheinungsbild haben können.
Unterscheidung zwischen Enchondrom und niedriggradigem Chondrosarkom:
Die Unterscheidung zwischen Enchondromen und niedriggradigen konventionellen Chondrosarkomen stellt eine häufige diagnostische Herausforderung dar, da sich die Läsionen sowohl histologisch als auch radiologisch sehr ähnlich sind. Es ist jedoch zu beachten, dass diese Unterscheidung in der Praxis umstritten sein kann, da beide klinisch und bildgebend engmaschig überwacht oder – bei Symptomatik – behandelt werden können.
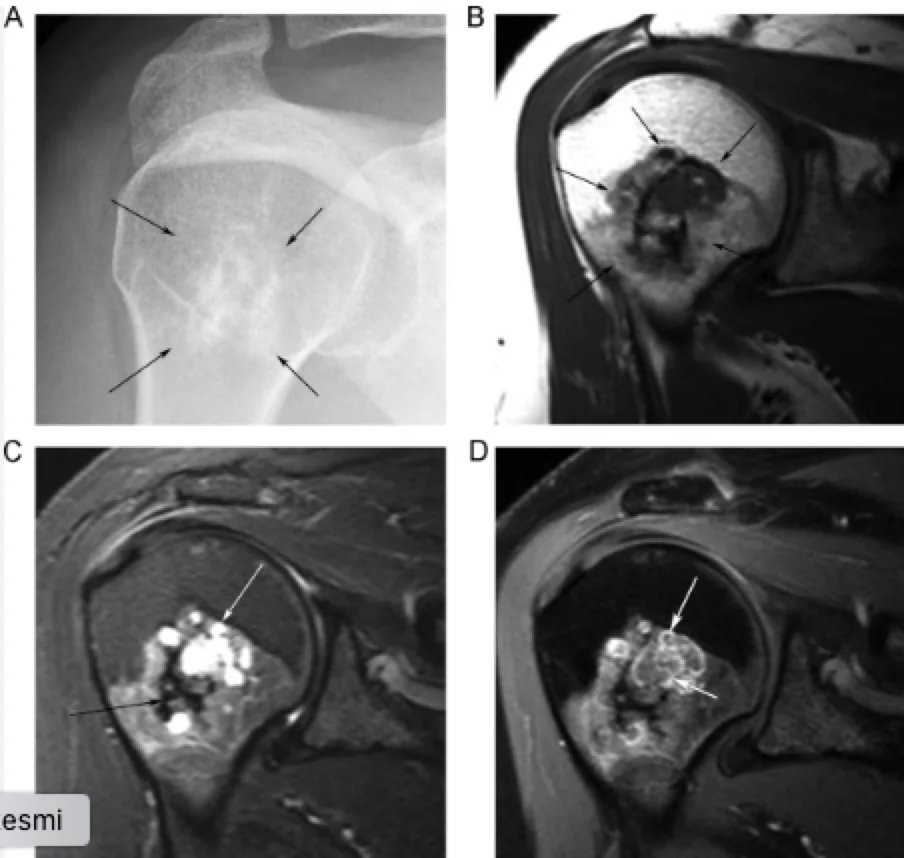
Radiologische Merkmale:
- Größe
Eine Läsionsgröße von über 5–6 cm spricht für ein Chondrosarkom.
- Durchbruch der Kortikalis
Wird bei 88 % der Chondrosarkome in langen Röhrenknochen beobachtet.
Kommt nur bei 8 % der Enchondrome vor.
- Tiefe endostale Fistel, die mehr als 2/3 der Kortikalisdicke umfasst
Wird bei 90 % der Chondrosarkome beobachtet.
Kommt nur bei 10 % der Enchondrome vor.
- Permeatives oder mottenfraßartiges Knochenbild
Wird bei hochgradigen Chondrosarkomen beobachtet.
- Weichteilmasse
Wird bei Enchondromen nicht beobachtet.
- Erhöhter Uptake in der Knochenszintigrafie
Wird bei 82 % der Chondrosarkome beobachtet.
Wird nur bei 21 % der Enchondrome beobachtet.
- Lokalisation
Hände und Füße sind seltene Lokalisationen für Chondrosarkome. Außerhalb von Händen und Füßen kommen Chondrosarkome häufiger vor als Enchondrome. Die Wirbelsäule, das Becken, das Kreuzbein und die Rippen sind Regionen, in denen Enchondrome selten beobachtet werden.
- Alter der Patient:innen
Enchondrome treten meist bei jungen Erwachsenen auf.
Chondrosarkome hingegen neigen dazu, bei Menschen mittleren Alters aufzutreten.
- Schmerzen
Chondrosarkome verursachen nahezu immer Schmerzen.
Enchondrome sind in der Regel schmerzlos – es sei denn, sie verursachen eine pathologische Fraktur.
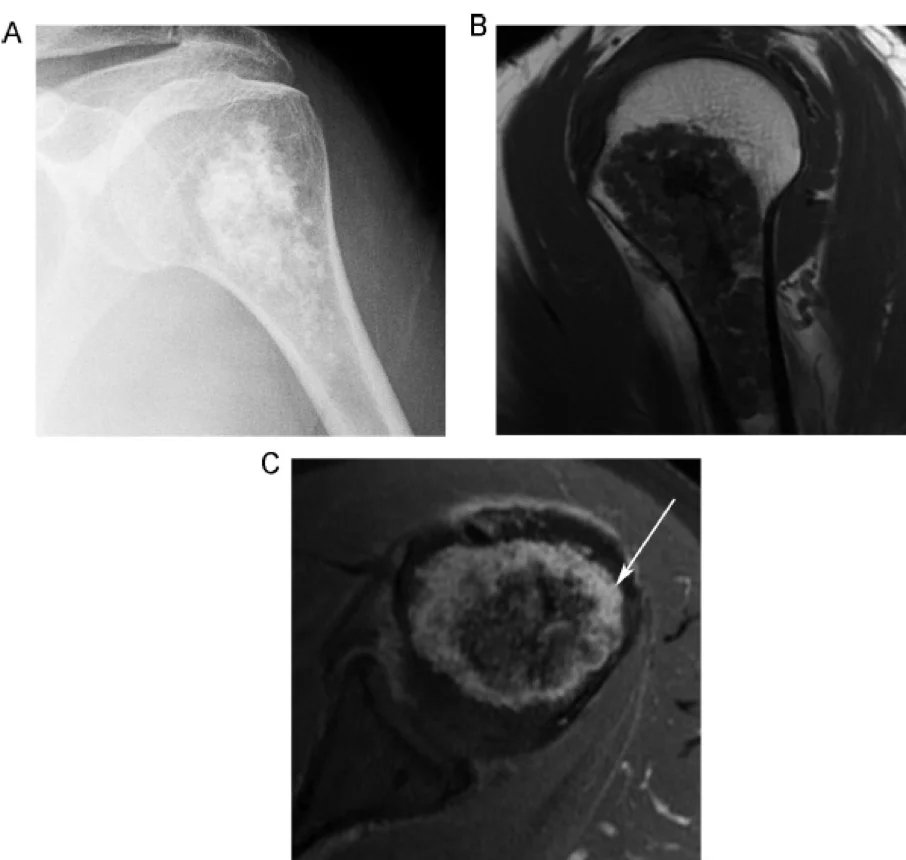
Behandlung des Enchondroms:
Da Enchondrome als Bone-RADS 1-Läsionen eingestuft werden, gelten sie typischerweise als gutartig und sollten unbehandelt bleiben, sofern sie keine Symptome verursachen. Die Mehrheit der Enchondrome bleibt asymptomatisch und erfordert keine Therapie. Pathologische Frakturen werden in der Regel durch Kürettage und Knochenaugmentation behandelt; zur Kontrolle von Heilung und Rückfall werden Röntgenkontrollen durchgeführt. Vor der Operation wird eine Biopsie entnommen. Die Rezidivrate liegt bei 2–15 % und kann auf eine maligne Transformation hindeuten.
Ist eine maligne Entartung wahrscheinlich, was in weniger als 5 % der Fälle vorkommt, erfolgt eine aggressivere Therapie.
